

Enhanced acetylation of ATP-citrate lyase promotes the progression of nonalcoholic fatty liver disease
- PMID: 31197036
- PMCID: PMC6682731
- DOI: 10.1074/jbc.RA119.008708
Enhanced acetylation of ATP-citrate lyase promotes the progression of nonalcoholic fatty liver disease
Abstract
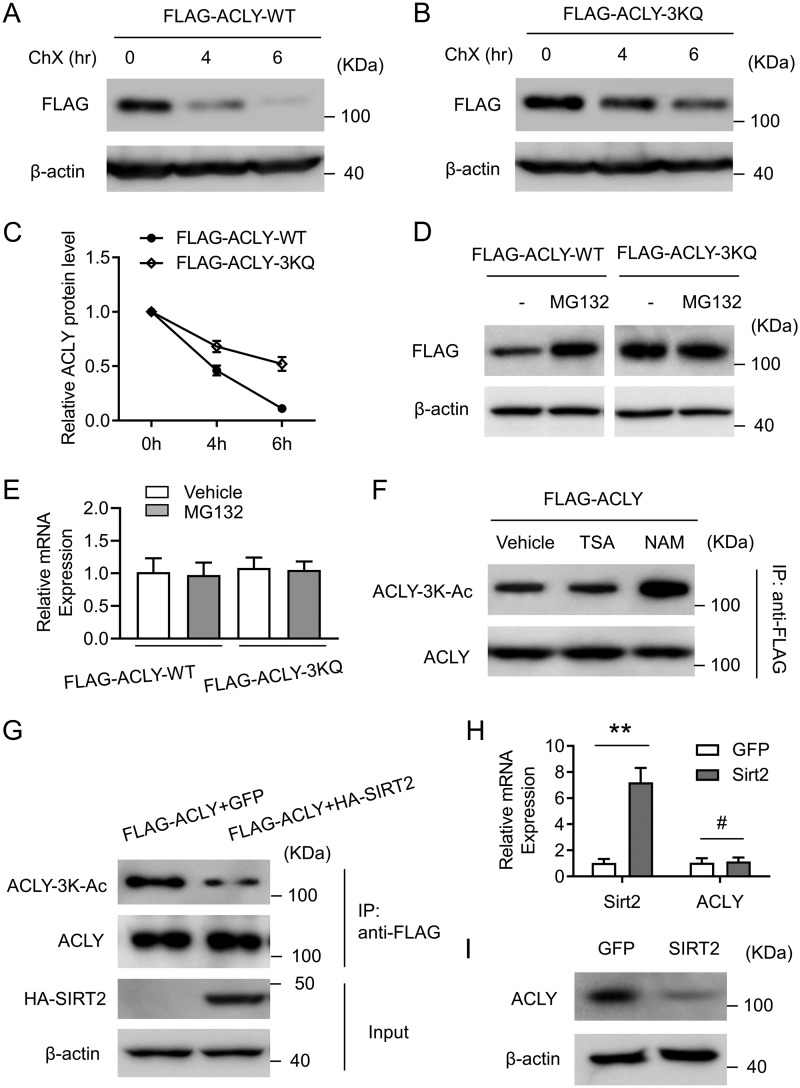
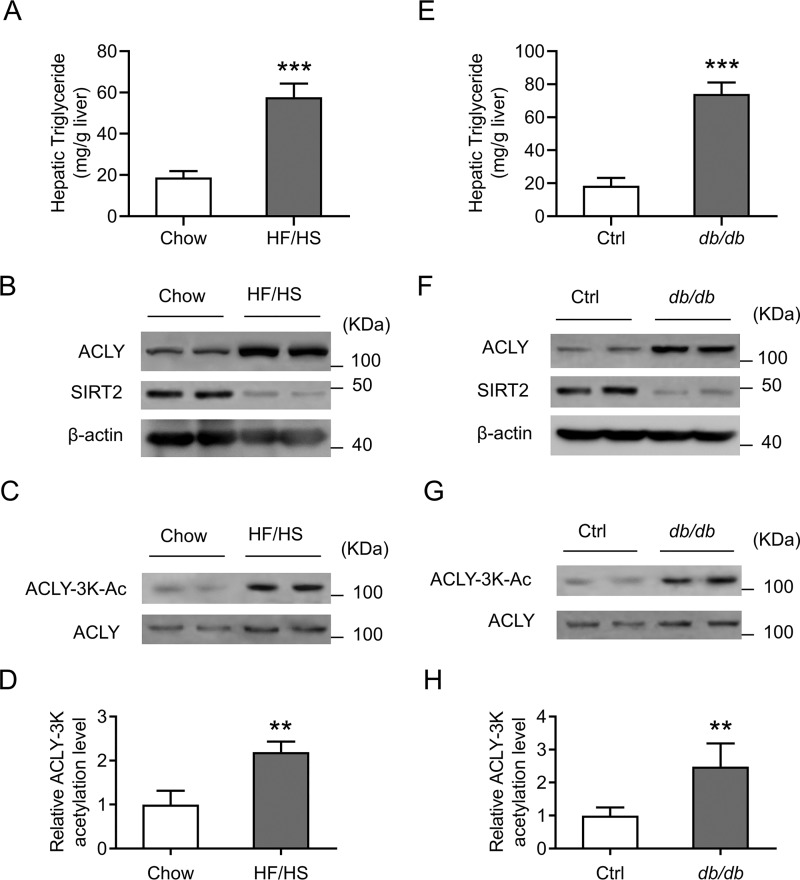
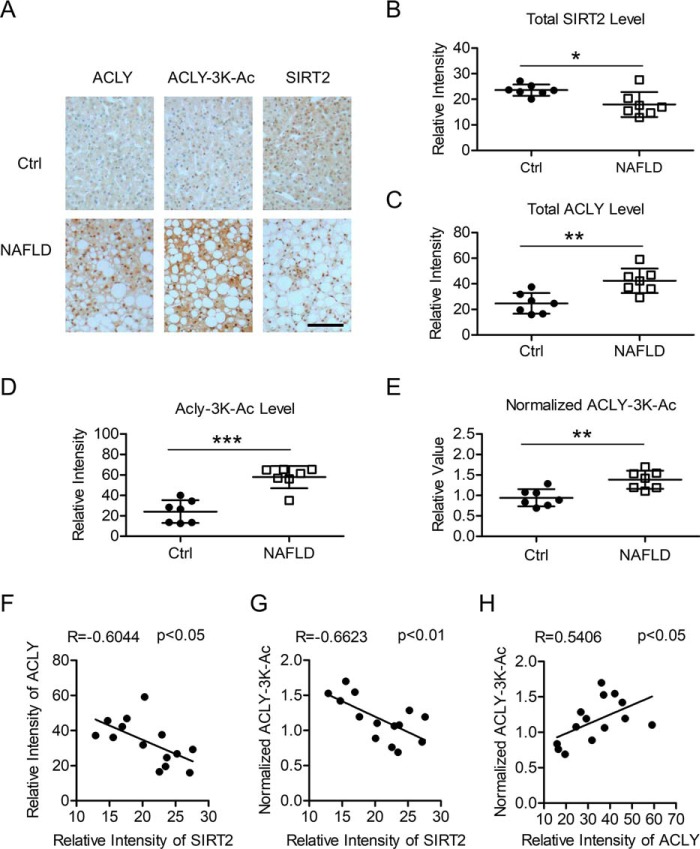
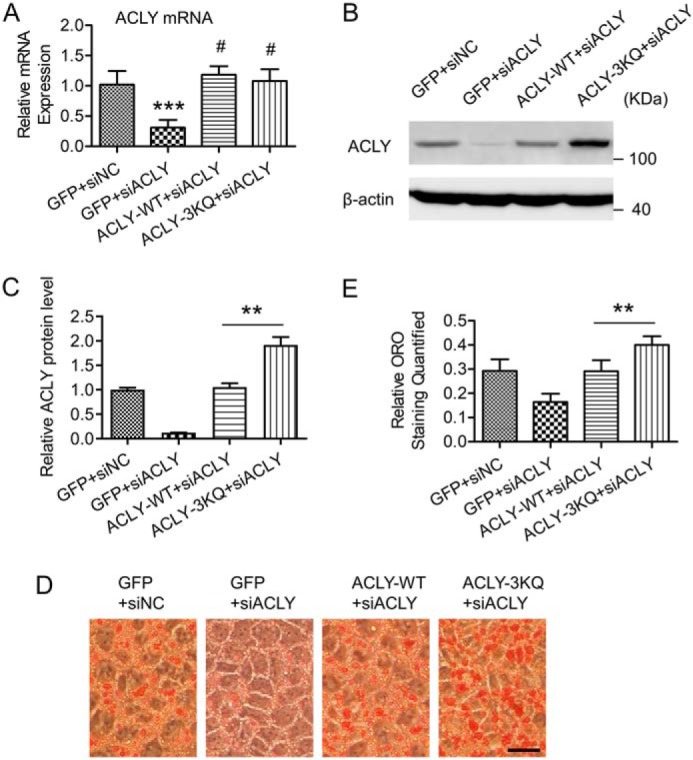
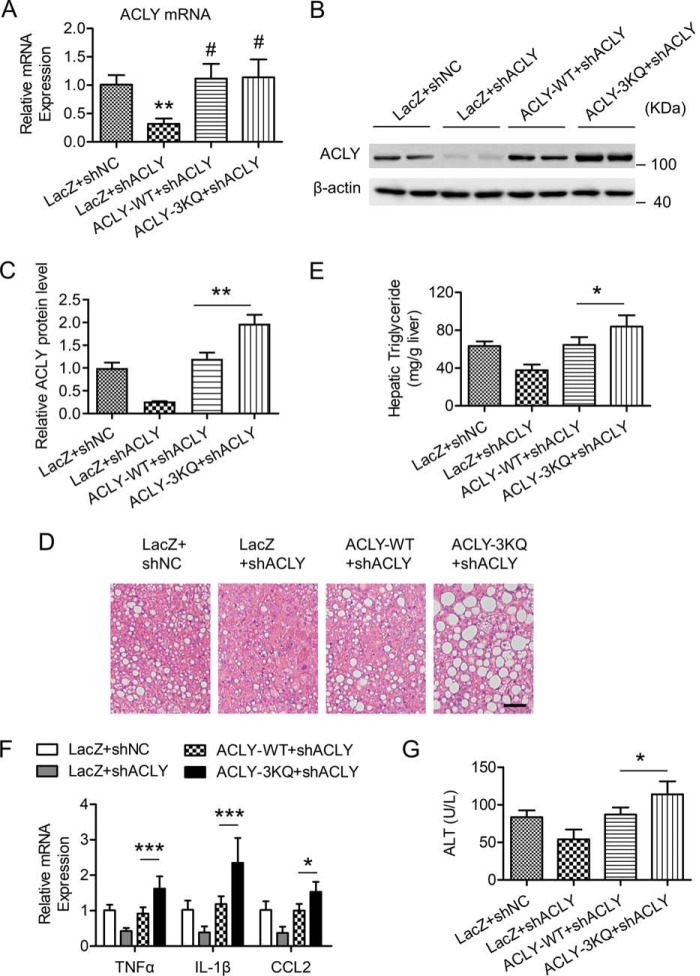
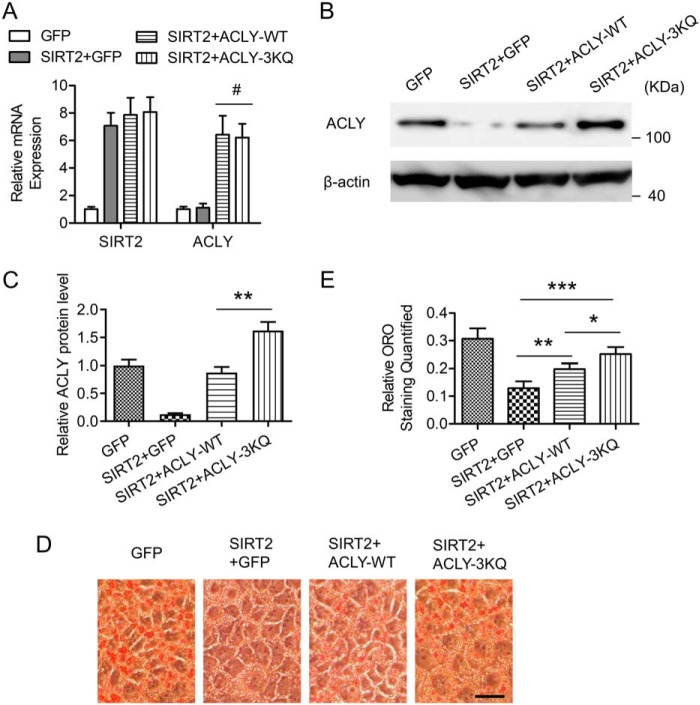
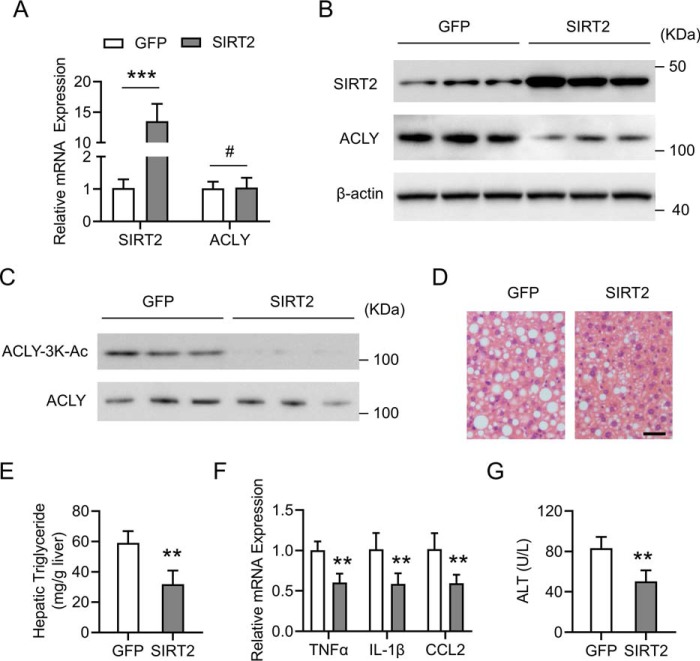
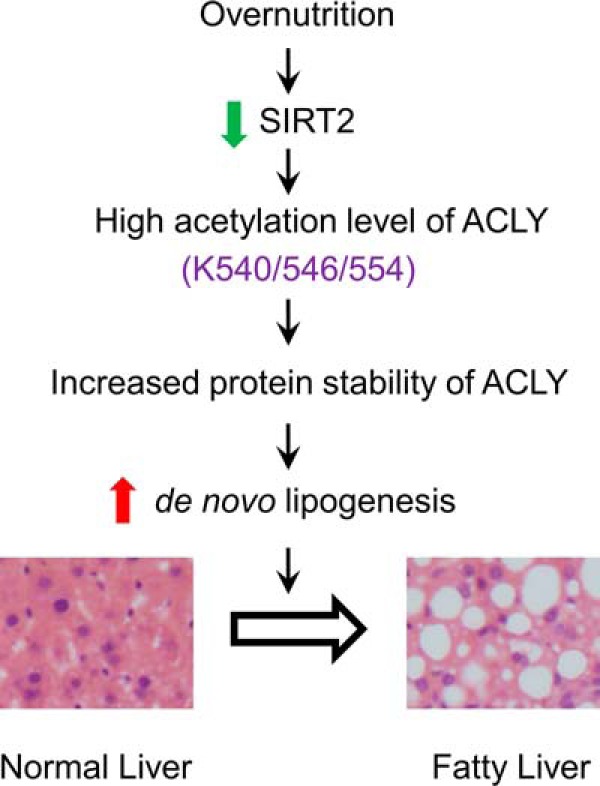
Hepatic steatosis is a hallmark of nonalcoholic fatty liver disease (NAFLD) and is promoted by dysregulated de novo lipogenesis. ATP-citrate lyase (ACLY) is a crucial lipogenic enzyme that is up-regulated in individuals with NAFLD. A previous study has shown that acetylation of ACLY at Lys-540, Lys-546, and Lys-554 (ACLY-3K) increases ACLY protein stability by antagonizing its ubiquitylation, thereby promoting lipid synthesis and cell proliferation in lung cancer cells. But the functional importance of this regulatory mechanism in other cellular or tissue contexts or under other pathophysiological conditions awaits further investigation. Here, we show that ACLY-3K acetylation also promotes ACLY protein stability in AML12 cells, a mouse hepatocyte cell line, and found that the deacetylase sirtuin 2 (SIRT2) deacetylates ACLY-3K and destabilizes ACLY in these cells. Of note, the livers of mice and humans with NAFLD had increased ACLY protein and ACLY-3K acetylation levels and decreased SIRT2 protein levels. Mimicking ACLY-3K acetylation by replacing the three lysines with three glutamines (ACLY-3KQ variant) promoted lipid accumulation both in high glucose-treated AML12 cells and in the livers of high-fat/high-sucrose (HF/HS) diet-fed mice. Moreover, overexpressing SIRT2 in AML12 cells inhibited lipid accumulation, which was more efficiently reversed by overexpressing the ACLY-3KQ variant than by overexpressing WT ACLY. Additionally, hepatic SIRT2 overexpression decreased ACLY-3K acetylation and its protein level and alleviated hepatic steatosis in HF/HS diet-fed mice. Our findings reveal a posttranscriptional mechanism underlying the up-regulation of hepatic ACLY in NAFLD and suggest that the SIRT2/ACLY axis is involved in NAFLD progression.
Keywords: ATP-citrate lyase (ACLY); acetylation; deacetylase; hepatic steatosis; hepatocyte; lipid metabolism; lipogenesis; metabolic disorder; nonalcoholic fatty liver disease (NAFLD); protein stability; sirtuin.
© 2019 Guo et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Marchesini G., and Babini M. (2006) Nonalcoholic fatty liver disease and the metabolic syndrome. Minerva Cardioangiol. 54, 229–239 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
