

CAM: an alignment-free method to recover phylogenies using codon aversion motifs
- PMID: 31198636
- PMCID: PMC6555396
- DOI: 10.7717/peerj.6984
CAM: an alignment-free method to recover phylogenies using codon aversion motifs
Abstract
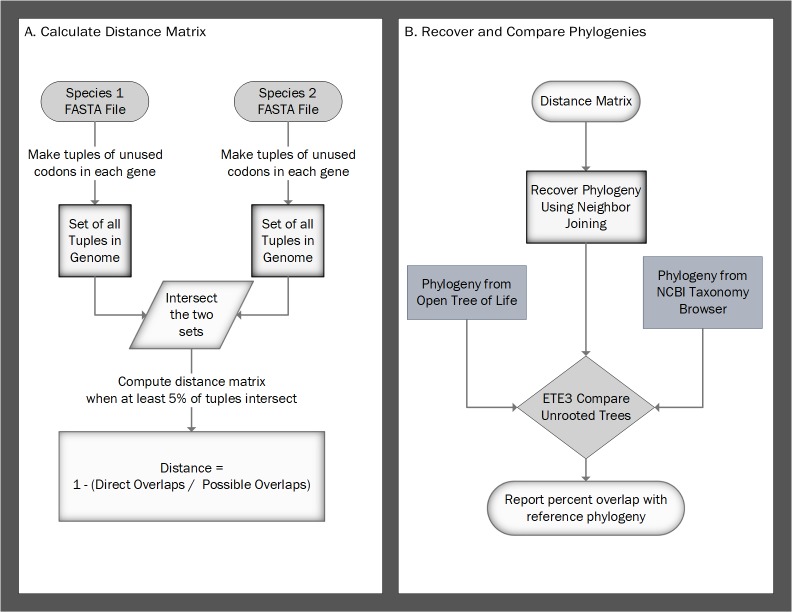
Background: Common phylogenomic approaches for recovering phylogenies are often time-consuming and require annotations for orthologous gene relationships that are not always available. In contrast, alignment-free phylogenomic approaches typically use structure and oligomer frequencies to calculate pairwise distances between species. We have developed an approach to quickly calculate distances between species based on codon aversion.
Methods: Utilizing a novel alignment-free character state, we present CAM, an alignment-free approach to recover phylogenies by comparing differences in codon aversion motifs (i.e., the set of unused codons within each gene) across all genes within a species. Synonymous codon usage is non-random and differs between organisms, between genes, and even within a single gene, and many genes do not use all possible codons. We report a comprehensive analysis of codon aversion within 229,742,339 genes from 23,428 species across all kingdoms of life, and we provide an alignment-free framework for its use in a phylogenetic construct. For each species, we first construct a set of codon aversion motifs spanning all genes within that species. We define the pairwise distance between two species, A and B, as one minus the number of shared codon aversion motifs divided by the total codon aversion motifs of the species, A or B, containing the fewest motifs. This approach allows us to calculate pairwise distances even when substantial differences in the number of genes or a high rate of divergence between species exists. Finally, we use neighbor-joining to recover phylogenies.
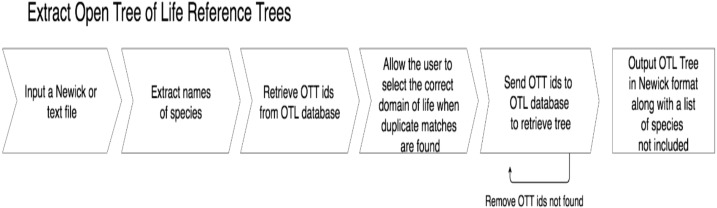
Results: Using the Open Tree of Life and NCBI Taxonomy Database as expected phylogenies, our approach compares well, recovering phylogenies that largely match expected trees and are comparable to trees recovered using maximum likelihood and other alignment-free approaches. Our technique is much faster than maximum likelihood and similar in accuracy to other alignment-free approaches. Therefore, we propose that codon aversion be considered a phylogenetically conserved character that may be used in future phylogenomic studies.
Availability: CAM, documentation, and test files are freely available on GitHub at https://github.com/ridgelab/cam.
Keywords: Alignment-free; Codon aversion; Codon usage bias; Maximum likelihood; Phylogenetics; Phylogenomics; Phylogeny; Systematics; Taxonomy; Tree of life.
Conflict of interest statement
The authors declare there are no competing interests.
Figures
Similar articles
-
Codon Pairs are Phylogenetically Conserved: A comprehensive analysis of codon pairing conservation across the Tree of Life.PLoS One. 2020 May 13;15(5):e0232260. doi: 10.1371/journal.pone.0232260. eCollection 2020. PLoS One. 2020. PMID: 32401752 Free PMC article.
-
Missing something? Codon aversion as a new character system in phylogenetics.Cladistics. 2017 Oct;33(5):545-556. doi: 10.1111/cla.12183. Epub 2017 Feb 3. Cladistics. 2017. PMID: 34706488
-
Codon use and aversion is largely phylogenetically conserved across the tree of life.Mol Phylogenet Evol. 2020 Mar;144:106697. doi: 10.1016/j.ympev.2019.106697. Epub 2019 Dec 2. Mol Phylogenet Evol. 2020. PMID: 31805345
-
A comprehensive analysis of the phylogenetic signal in ramp sequences in 211 vertebrates.Sci Rep. 2021 Jan 12;11(1):622. doi: 10.1038/s41598-020-78803-3. Sci Rep. 2021. PMID: 33436653 Free PMC article.
-
Differential expression of the three independent CaM genes coding for an identical protein: Potential relevance of distinct mRNA stability by different codon usage.Cell Calcium. 2022 Nov;107:102656. doi: 10.1016/j.ceca.2022.102656. Epub 2022 Oct 8. Cell Calcium. 2022. PMID: 36252447 Review.
Cited by
-
Ramp Sequence May Explain Synonymous Variant Association with Alzheimer's Disease in the Paired Immunoglobulin-like Type 2 Receptor Alpha (PILRA).Biomedicines. 2025 Mar 18;13(3):739. doi: 10.3390/biomedicines13030739. Biomedicines. 2025. PMID: 40149715 Free PMC article.
-
Plastome evolution of Aeonium and Monanthes (Crassulaceae): insights into the variation of plastomic tRNAs, and the patterns of codon usage and aversion.Planta. 2022 Jul 9;256(2):35. doi: 10.1007/s00425-022-03950-y. Planta. 2022. PMID: 35809200
-
CUBAP: an interactive web portal for analyzing codon usage biases across populations.Nucleic Acids Res. 2020 Nov 4;48(19):11030-11039. doi: 10.1093/nar/gkaa863. Nucleic Acids Res. 2020. PMID: 33045750 Free PMC article.
-
Ten Plastomes of Crassula (Crassulaceae) and Phylogenetic Implications.Biology (Basel). 2022 Dec 7;11(12):1779. doi: 10.3390/biology11121779. Biology (Basel). 2022. PMID: 36552287 Free PMC article.
-
Codon Pairs are Phylogenetically Conserved: A comprehensive analysis of codon pairing conservation across the Tree of Life.PLoS One. 2020 May 13;15(5):e0232260. doi: 10.1371/journal.pone.0232260. eCollection 2020. PLoS One. 2020. PMID: 32401752 Free PMC article.
References
LinkOut - more resources
Full Text Sources
