

Recombination-Aware Phylogenomics Reveals the Structured Genomic Landscape of Hybridizing Cat Species
- PMID: 31198971
- PMCID: PMC6759079
- DOI: 10.1093/molbev/msz139
Recombination-Aware Phylogenomics Reveals the Structured Genomic Landscape of Hybridizing Cat Species
Abstract
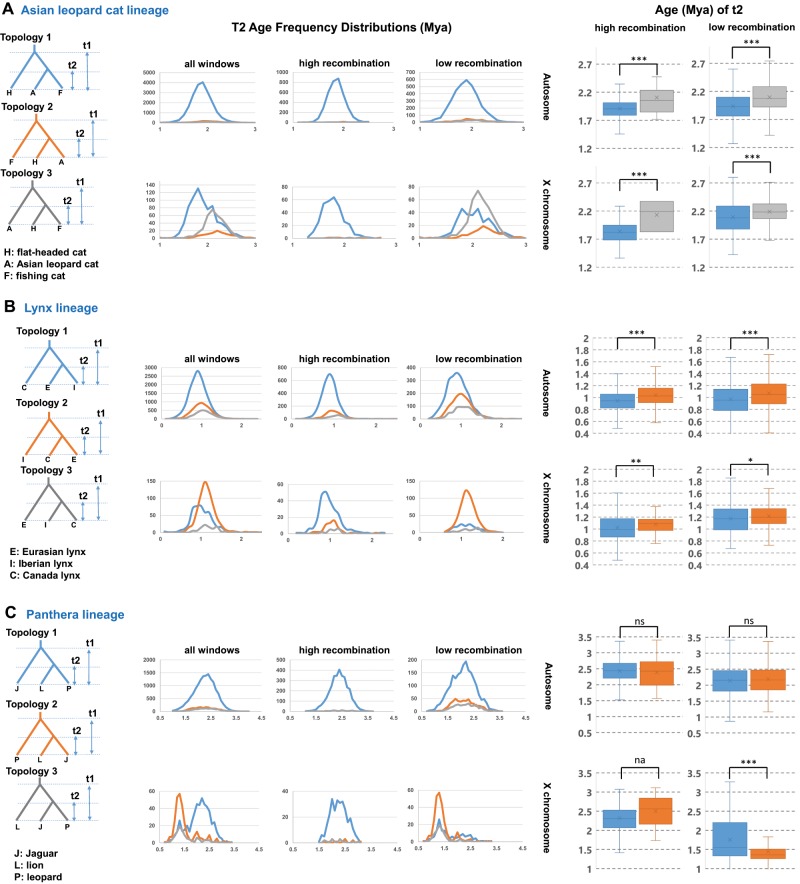
Current phylogenomic approaches implicitly assume that the predominant phylogenetic signal within a genome reflects the true evolutionary history of organisms, without assessing the confounding effects of postspeciation gene flow that can produce a mosaic of phylogenetic signals that interact with recombinational variation. Here, we tested the validity of this assumption with a phylogenomic analysis of 27 species of the cat family, assessing local effects of recombination rate on species tree inference and divergence time estimation across their genomes. We found that the prevailing phylogenetic signal within the autosomes is not always representative of the most probable speciation history, due to ancient hybridization throughout felid evolution. Instead, phylogenetic signal was concentrated within regions of low recombination, and notably enriched within large X chromosome recombination cold spots that exhibited recurrent patterns of strong genetic differentiation and selective sweeps across mammalian orders. By contrast, regions of high recombination were enriched for signatures of ancient gene flow, and these sequences inflated crown-lineage divergence times by ∼40%. We conclude that existing phylogenomic approaches to infer the Tree of Life may be highly misleading without considering the genomic architecture of phylogenetic signal relative to recombination rate and its interplay with historical hybridization.
Keywords: Felidae; X chromosome; hybridization; phylogenomics; recombination.
© The Author(s) 2019. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures







References
-
- Ai H, Fang X, Yang B, Huang Z, Chen H, Mao L, Zhang F, Zhang L, Cui L, He W.. 2015. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat Genet. 473:217–225. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
