

CXCL16/CXCR6 is involved in LPS-induced acute lung injury via P38 signalling
- PMID: 31199046
- PMCID: PMC6653424
- DOI: 10.1111/jcmm.14419
CXCL16/CXCR6 is involved in LPS-induced acute lung injury via P38 signalling
Abstract
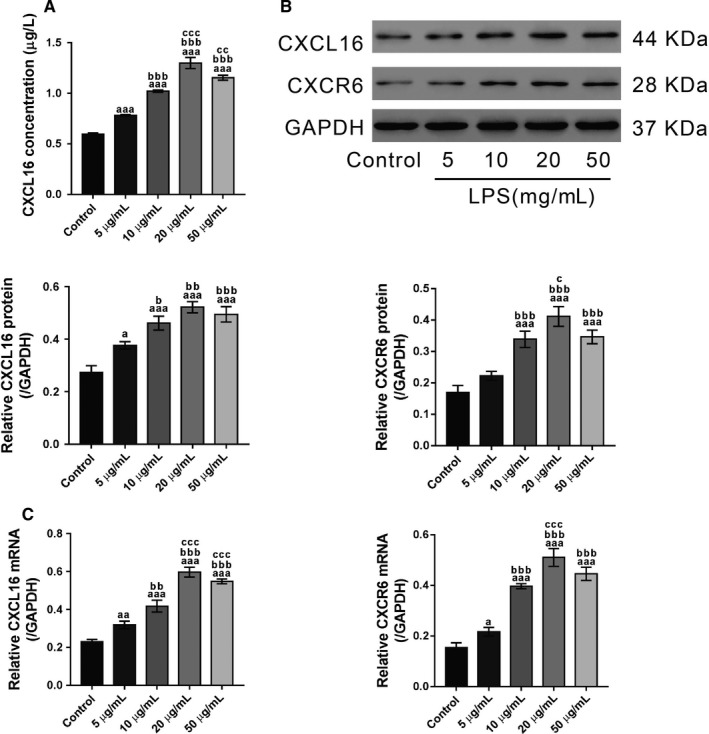
Although several chemokines play key roles in the pathogenesis of acute lung injury (ALI), the roles of chemokine (C-X-C motif) ligand 16 (CXCL16) and its receptor C-X-C chemokine receptor type 6 (CXCR6) in ALI pathogenesis remain to be elucidated. The mRNA and protein expression of CXCL16 and CXCR6 was detected after lipopolysaccharide (LPS) stimulation with or without treatment with the nuclear factor-κB (NF-κB) inhibitor pyrrolidine dithiocarbamate (PDTC). Lung injury induced by LPS was evaluated in CXCR6 knockout mice. CXCL16 level was elevated in the serum of ALI patients (n = 20) compared with healthy controls (n = 30). CXCL16 treatment (50, 100, and 200 ng/mL) in 16HBE cells significantly decreased the epithelial barrier integrity and E-cadherin expression, and increased CXCR6 expression, reactive oxygen species (ROS) production, and p38 phosphorylation. Knockdown of CXCR6 or treatment with the p38 inhibitor SB203580 abolished the effects of CXCL16. Moreover, treatment of 16HBE cells with LPS (5, 10, 20 and 50 μg/mL) significantly increased CXCL16 release as well as the mRNA and protein levels of CXCL16 and CXCR6. The effects of LPS treatment (20 μg/mL) were abolished by treatment with PDTC. The results of the luciferase assay further demonstrated that PDTC treatment markedly inhibited the activity of the CXCL16 promoter. In conclusion, CXCL16, whose transcription was enhanced by LPS, may be involved in ROS production, epithelial barrier dysfunction and E-cadherin down-regulation via p38 signalling, thus contributing to the pathogenesis of ALI. Importantly, CXCR6 knockout or inhibition of p38 signalling may protect mice from LPS-induced lung injury by decreasing E-cadherin expression.
Keywords: 16HBE; CXCL16; NF-κB; acute lung injury; p38 signal.
© 2019 The Authors. Journal of Cellular and Molecular Medicine published by John Wiley & Sons Ltd and Foundation for Cellular and Molecular Medicine.
Conflict of interest statement
The authors declare no competing interests.
Figures





Similar articles
-
Functional role of endothelial CXCL16/CXCR6-platelet-leucocyte axis in angiotensin II-associated metabolic disorders.Cardiovasc Res. 2018 Nov 1;114(13):1764-1775. doi: 10.1093/cvr/cvy135. Cardiovasc Res. 2018. PMID: 29800106
-
MiR-625-5p is a potential therapeutic target in sepsis by regulating CXCL16/CXCR6 axis and endothelial barrier.Int Immunopharmacol. 2024 Aug 20;137:112508. doi: 10.1016/j.intimp.2024.112508. Epub 2024 Jun 17. Int Immunopharmacol. 2024. PMID: 38889512
-
CXCL16/CXCR6 axis promotes bleomycin-induced fibrotic process in MRC-5 cells via the PI3K/AKT/FOXO3a pathway.Int Immunopharmacol. 2020 Apr;81:106035. doi: 10.1016/j.intimp.2019.106035. Epub 2019 Nov 19. Int Immunopharmacol. 2020. PMID: 31753588
-
The role of the CXCR6/CXCL16 axis in the pathogenesis of fibrotic disease.Int Immunopharmacol. 2024 May 10;132:112015. doi: 10.1016/j.intimp.2024.112015. Epub 2024 Apr 11. Int Immunopharmacol. 2024. PMID: 38608478 Review.
-
The role of CXCL16 in atherosclerosis: from mechanisms to therapy.Front Immunol. 2025 May 26;16:1555438. doi: 10.3389/fimmu.2025.1555438. eCollection 2025. Front Immunol. 2025. PMID: 40491927 Free PMC article. Review.
Cited by
-
Exosomes derived from dental pulp stem cells accelerate cutaneous wound healing by enhancing angiogenesis via the Cdc42/p38 MAPK pathway.Int J Mol Med. 2022 Dec;50(6):143. doi: 10.3892/ijmm.2022.5199. Epub 2022 Nov 2. Int J Mol Med. 2022. PMID: 36321793 Free PMC article.
-
The Emerging Roles of Tripartite Motif Proteins (TRIMs) in Acute Lung Injury.J Immunol Res. 2021 Oct 19;2021:1007126. doi: 10.1155/2021/1007126. eCollection 2021. J Immunol Res. 2021. PMID: 34712740 Free PMC article. Review.
-
CCL25 Inhibition Alleviates Sepsis-Induced Acute Lung Injury and Inflammation.Infect Drug Resist. 2022 Jun 25;15:3309-3321. doi: 10.2147/IDR.S352544. eCollection 2022. Infect Drug Resist. 2022. PMID: 35782530 Free PMC article.
-
Genetic and epigenetic factors associated with increased severity of Covid-19.Cell Biol Int. 2021 Jun;45(6):1158-1174. doi: 10.1002/cbin.11572. Epub 2021 Mar 1. Cell Biol Int. 2021. PMID: 33590936 Free PMC article. Review.
-
Pien Tze Huang alleviates Concanavalin A-induced autoimmune hepatitis by regulating intestinal microbiota and memory regulatory T cells.World J Gastroenterol. 2023 Dec 7;29(45):5988-6016. doi: 10.3748/wjg.v29.i45.5988. World J Gastroenterol. 2023. PMID: 38130997 Free PMC article.
References
-
- Bernard GR, Artigas A, Brigham KL, et al. The American‐European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149(3):818‐824. - PubMed
-
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334‐1349. - PubMed
-
- Chow CW, Herrera AM, Suzuki T, Downey GP. Oxidative stress and acute lung injury. Am J Respir Cell Mol Biol. 2003;29:427‐431. - PubMed
-
- Baldwin SR, Simon RH, Grum CM, Ketai LH, Boxer LA, Devall LJ. Oxidant activity in expired breath of patients with adult respiratory distress syndrome. Lancet. 1986;327:11‐14. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
