

Transition State Asymmetry in C-H Bond Cleavage by Proton-Coupled Electron Transfer
- PMID: 31199137
- PMCID: PMC7241443
- DOI: 10.1021/jacs.9b04303
Transition State Asymmetry in C-H Bond Cleavage by Proton-Coupled Electron Transfer
Abstract
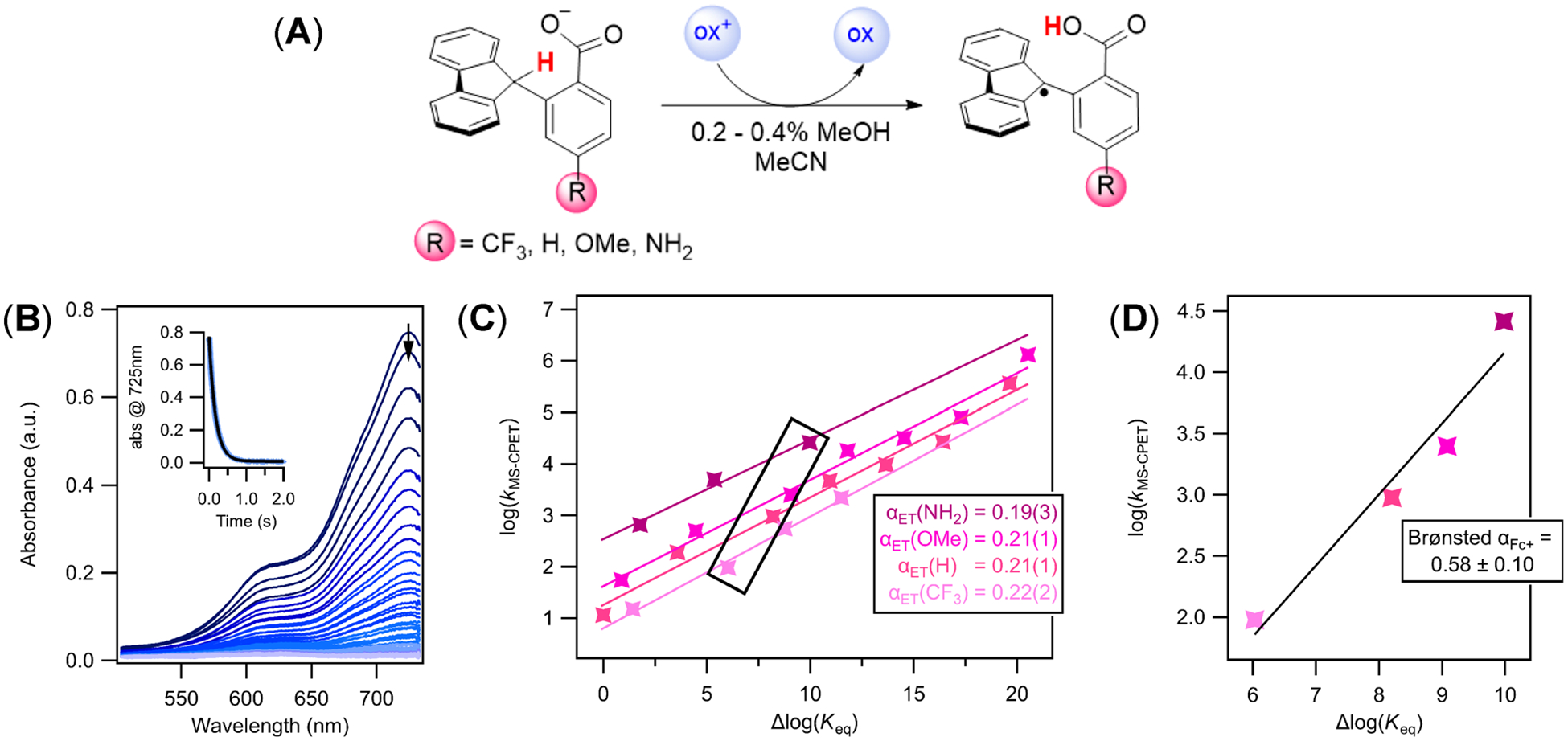
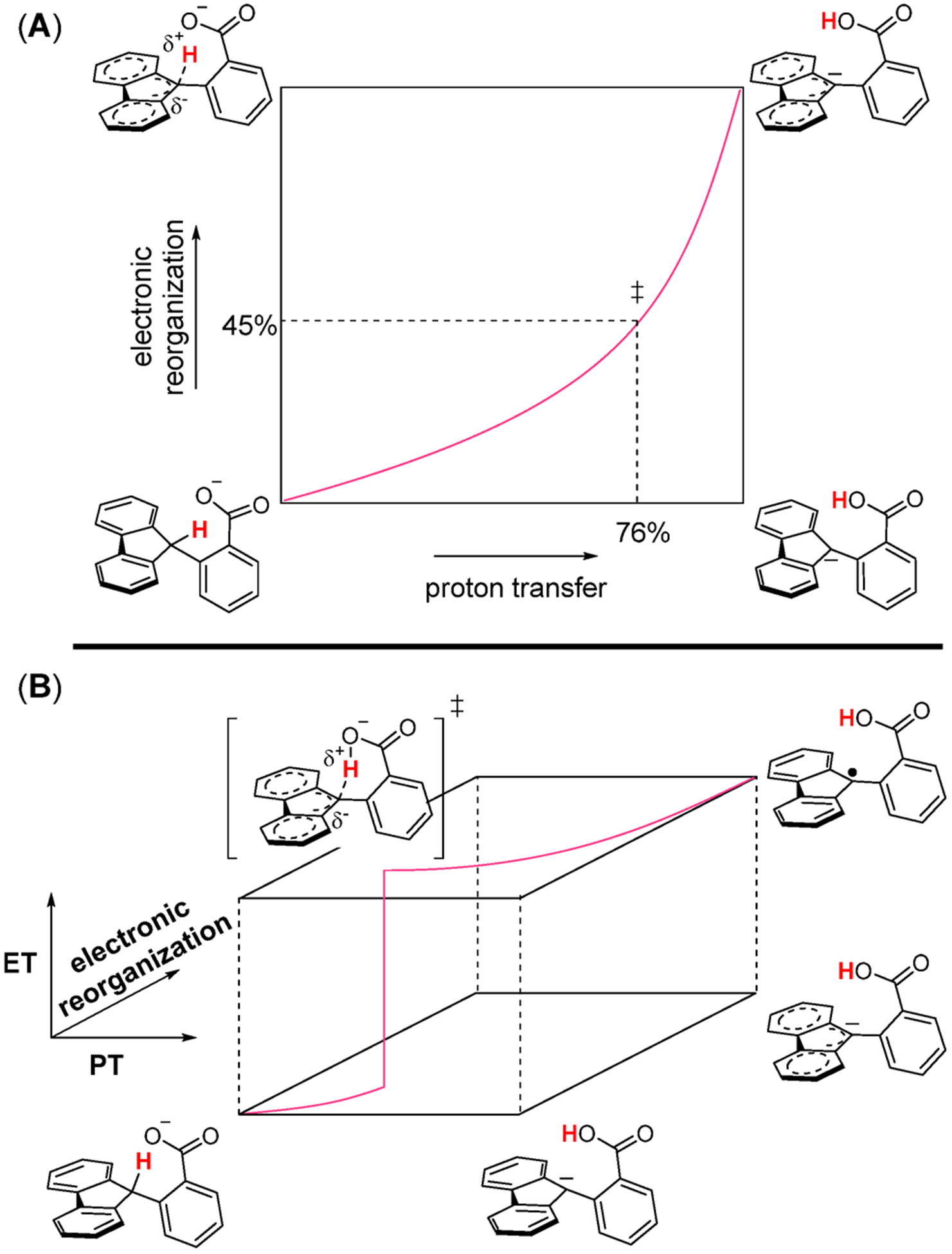
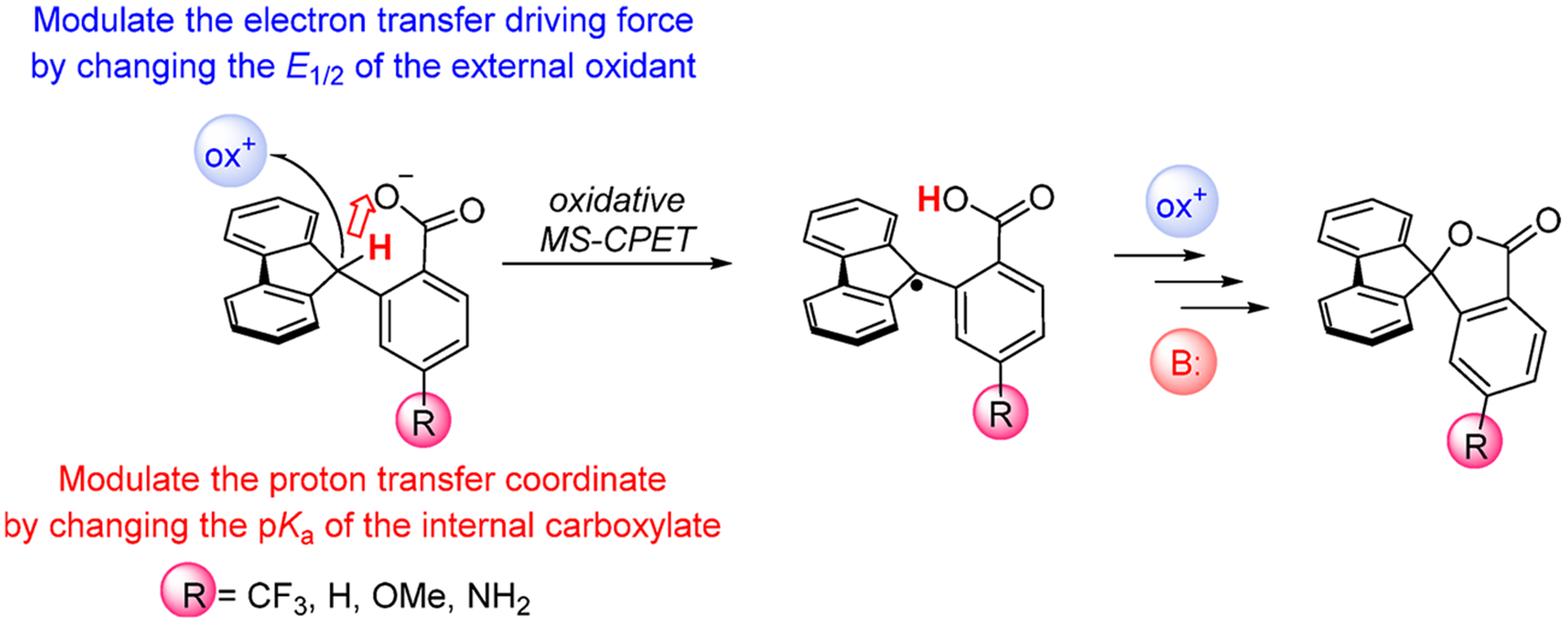
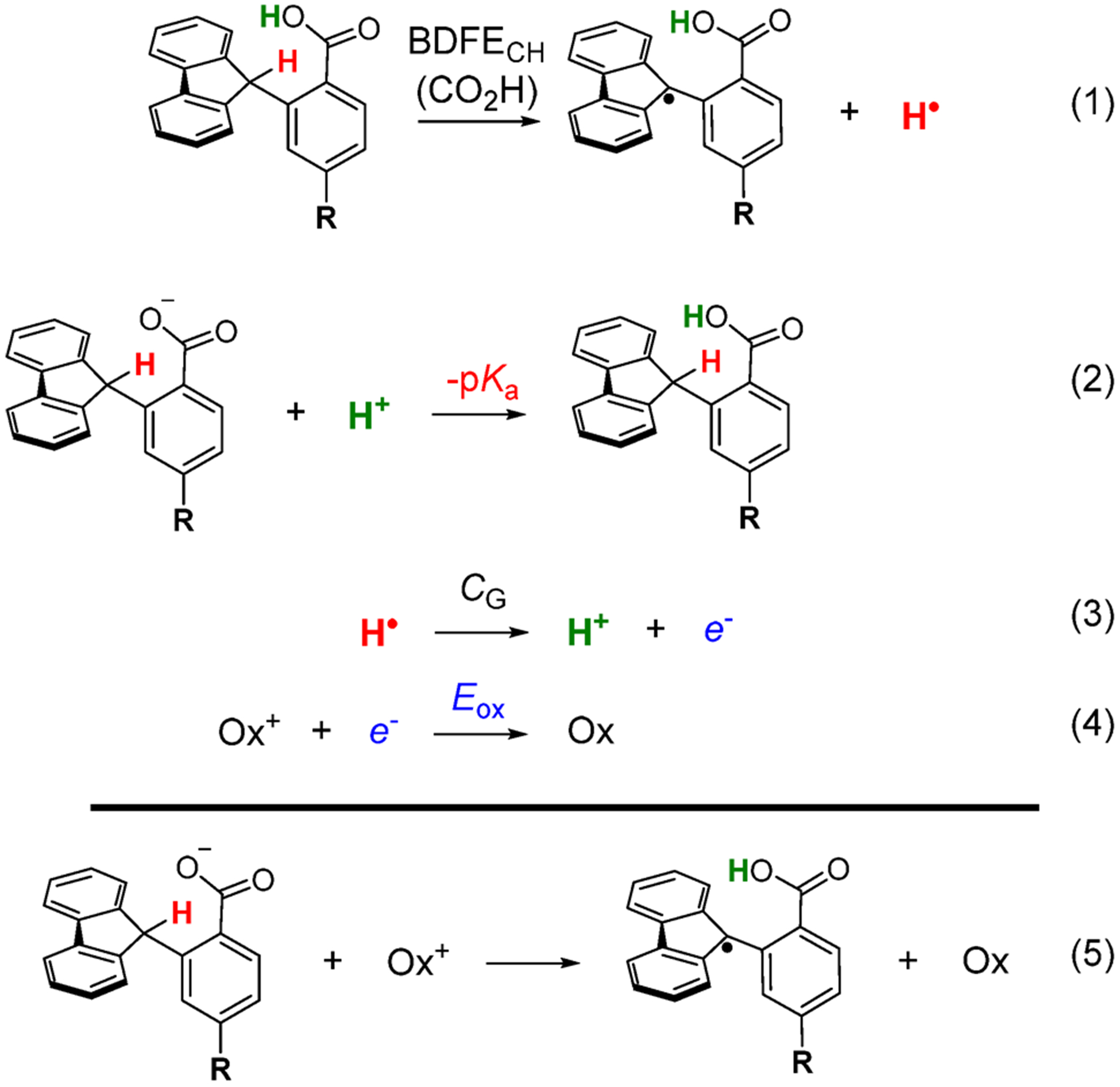
The selective transformation of C-H bonds is a longstanding challenge in modern chemistry. A recent report details C-H oxidation via multiple-site concerted proton-electron transfer (MS-CPET), where the proton and electron in the C-H bond are transferred to separate sites. Reactivity at a specific C-H bond was achieved by appropriate positioning of an internal benzoate base. Here, we extend that report to reactions of a series of molecules with differently substituted fluorenyl-benzoates and varying outer-sphere oxidants. These results probe the fundamental rate versus driving force relationships in this MS-CPET reaction at carbon by separately modulating the driving force for the proton and electron transfer components. The rate constants depend strongly on the pKa of the internal base, but depend much less on the nature of the outer-sphere oxidant. These observations suggest that the transition states for these reactions are imbalanced. Density functional theory (DFT) was used to generate an internal reaction coordinate, which qualitatively reproduced the experimental observation of a transition state imbalance. Thus, in this system, homolytic C-H bond cleavage involves concerted but asynchronous transfer of the H+ and e-. The nature of this transfer has implications for synthetic methodology and biological systems.
Conflict of interest statement
The authors declare no competing financial interest.
Figures

References
-
- Hartwig JF Evolution of C–H bond functionalization from methane to methodology. J. Am. Chem. Soc 2016, 138, 2. - PMC - PubMed
- Labinger JA; Bercaw JE Understanding and exploiting C–H bond activation. Nature 2002, 417, 507. - PubMed
- He J; Wasa M; Chan KSL; Shao Q; Yu J-Q Palladium-Catalyzed Transformations of Alkyl C–H Bonds. Chem. Rev 2017, 117, 8754. - PMC - PubMed
- Crabtree RH Alkane C–H activation and functionalization with homogeneous transition metal catalysts: A century of progress—A new millennium in prospect. J. Chem. Soc., Dalton Trans 2001, 2437.
- Davies HML; Morton D Recent Advances in C–H Functionalization. J. Org. Chem 2016, 81, 343. - PubMed
- Goldberg KI; Goldman AS Activation and Functionalization of CH Bonds; American Chemical Society: Washington, DC, 2004; Vol. 885.
- Shul’pin GB Selectivity enhancement in functionalization of C–H bonds: A review. Org. Biomol. Chem 2010, 8, 4217. - PubMed
-
- Kochi JK Free Radicals; Wiley: New York, 1973; Vol. 2
- Perkins M Free Radical Chemistry; Ellis Horwood: New York, 1994.
- Burton G; Ingold KU Vitamin E: application of the principles of physical organic chemistry to the exploration of its structure and function. Acc. Chem. Res 1986, 19, 194.
- Gutteridge JM; Halliwell B The measurement and mechanism of lipid peroxidation in biological systems. Trends Biochem. Sci 1990, 15, 129. - PubMed
- Olah GA; Prakash GS Hydrocarbon Chemistry; John Wiley & Sons: New York, 1995.
-
- Snider BB Manganese(III)-Based Oxidative Free-Radical Cyclizations. Chem. Rev 1996, 96, 339. - PubMed
- Mayer JM Hydrogen atom abstraction by metal–oxo complexes: understanding the analogy with organic radical reactions. Acc. Chem. Res 1998, 31, 441.
-
- Reece SY; Nocera DG Proton-Coupled Electron Transfer in Biology: Results from Synergistic Studies in Natural and Model Systems. Annu. Rev. Biochem 2009, 78, 673. - PMC - PubMed
- Jasniewski AJ; Que L Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev 2018, 118, 2554. - PMC - PubMed
- Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev 2014, 114, 3659. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
