

Decrypting protein surfaces by combining evolution, geometry, and molecular docking
- PMID: 31199528
- PMCID: PMC6852240
- DOI: 10.1002/prot.25757
Decrypting protein surfaces by combining evolution, geometry, and molecular docking
Abstract
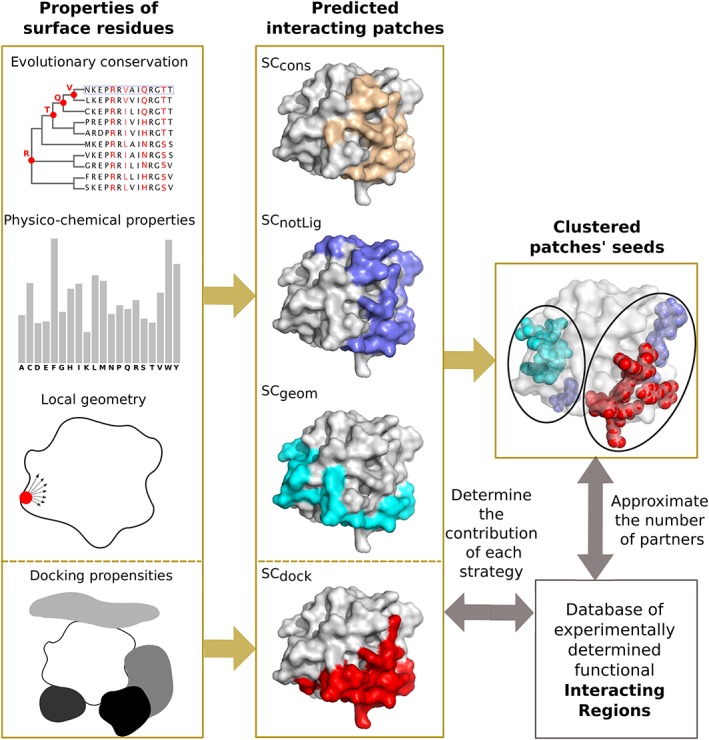
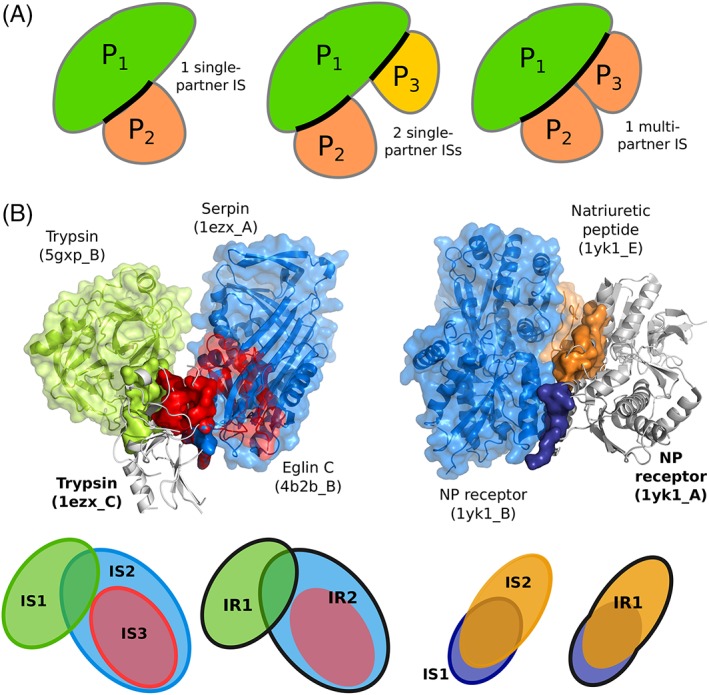
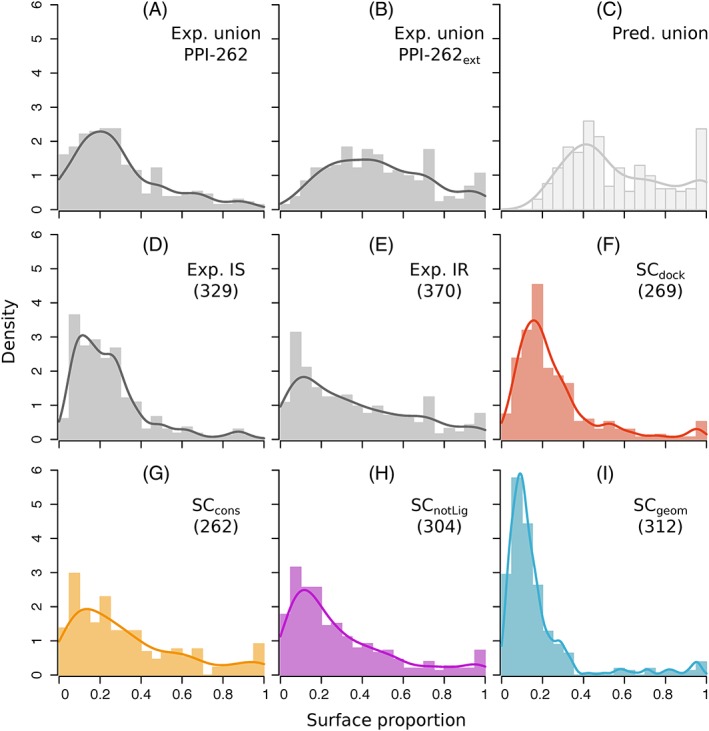
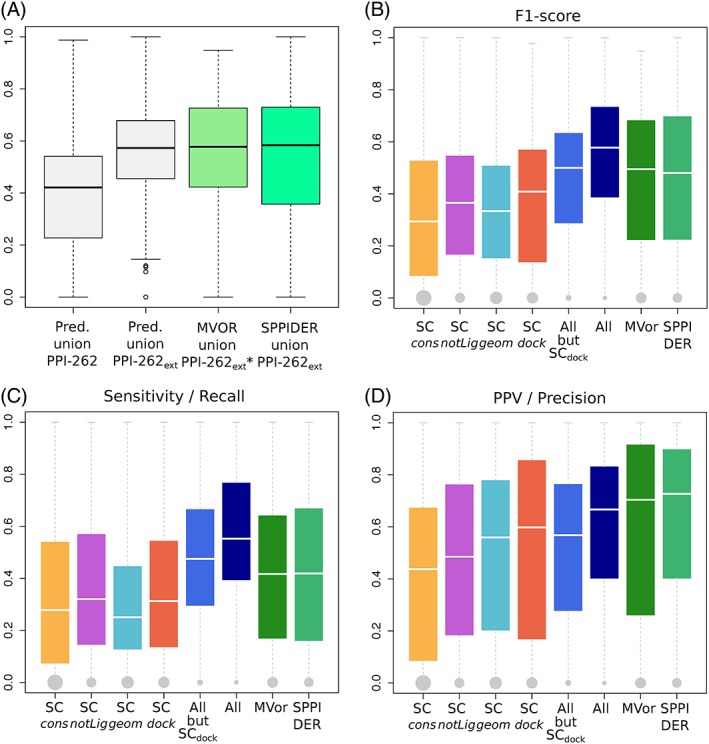
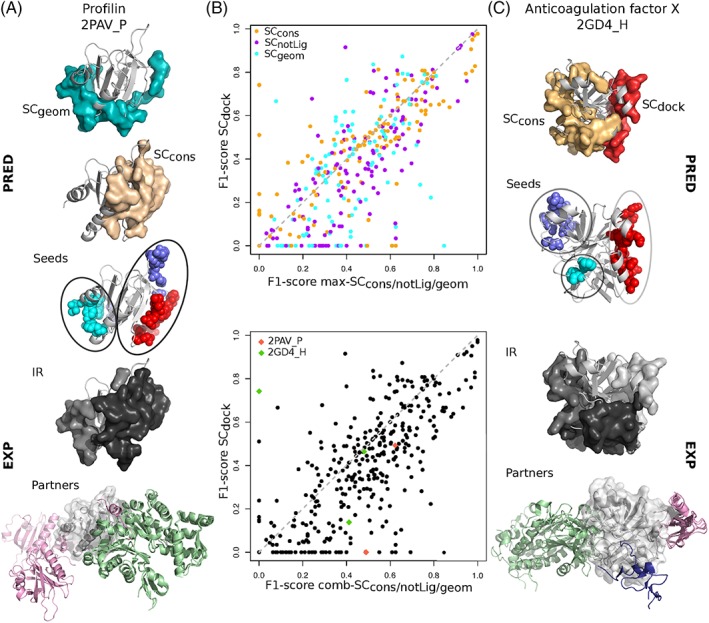
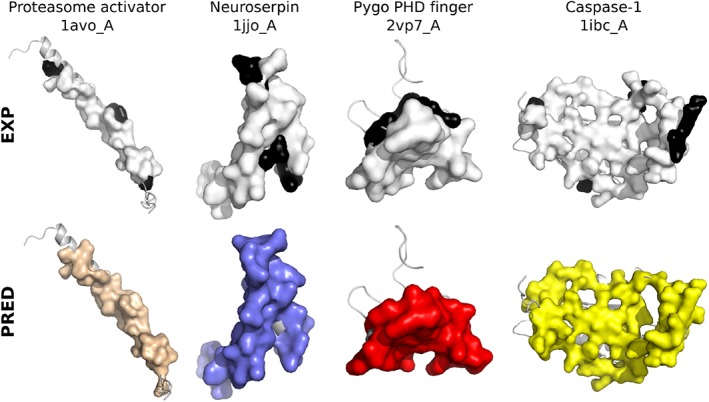
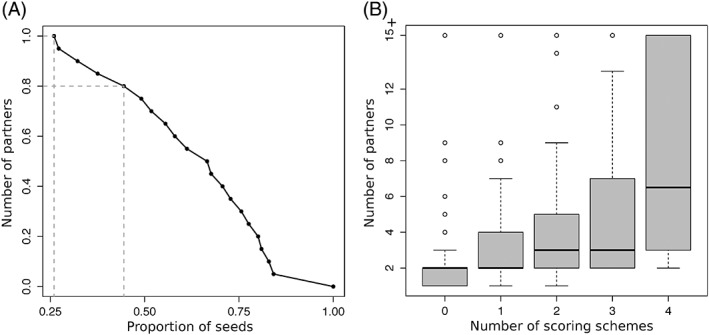
The growing body of experimental and computational data describing how proteins interact with each other has emphasized the multiplicity of protein interactions and the complexity underlying protein surface usage and deformability. In this work, we propose new concepts and methods toward deciphering such complexity. We introduce the notion of interacting region to account for the multiple usage of a protein's surface residues by several partners and for the variability of protein interfaces coming from molecular flexibility. We predict interacting patches by crossing evolutionary, physicochemical and geometrical properties of the protein surface with information coming from complete cross-docking (CC-D) simulations. We show that our predictions match well interacting regions and that the different sources of information are complementary. We further propose an indicator of whether a protein has a few or many partners. Our prediction strategies are implemented in the dynJET2 algorithm and assessed on a new dataset of 262 protein on which we performed CC-D. The code and the data are available at: http://www.lcqb.upmc.fr/dynJET2/.
Keywords: binding site; complete cross-docking; evolutionary conservation; interface prediction; protein-protein interaction.
© 2019 The Authors. Proteins: Structure, Function, and Bioinformatics published by Wiley Periodicals, Inc.
Conflict of interest statement
The authors have no conflict of interest to declare.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
