

The gene regulatory basis of genetic compensation during neural crest induction
- PMID: 31199790
- PMCID: PMC6594659
- DOI: 10.1371/journal.pgen.1008213
The gene regulatory basis of genetic compensation during neural crest induction
Abstract
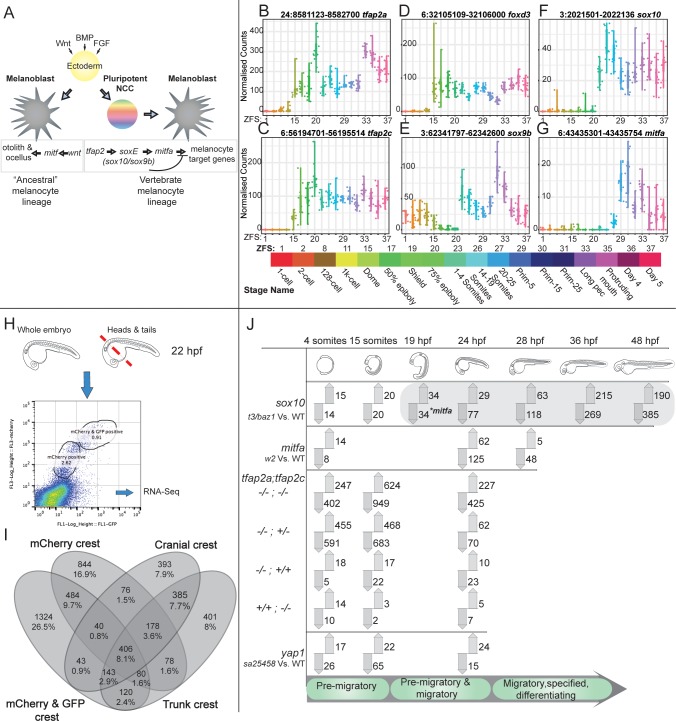
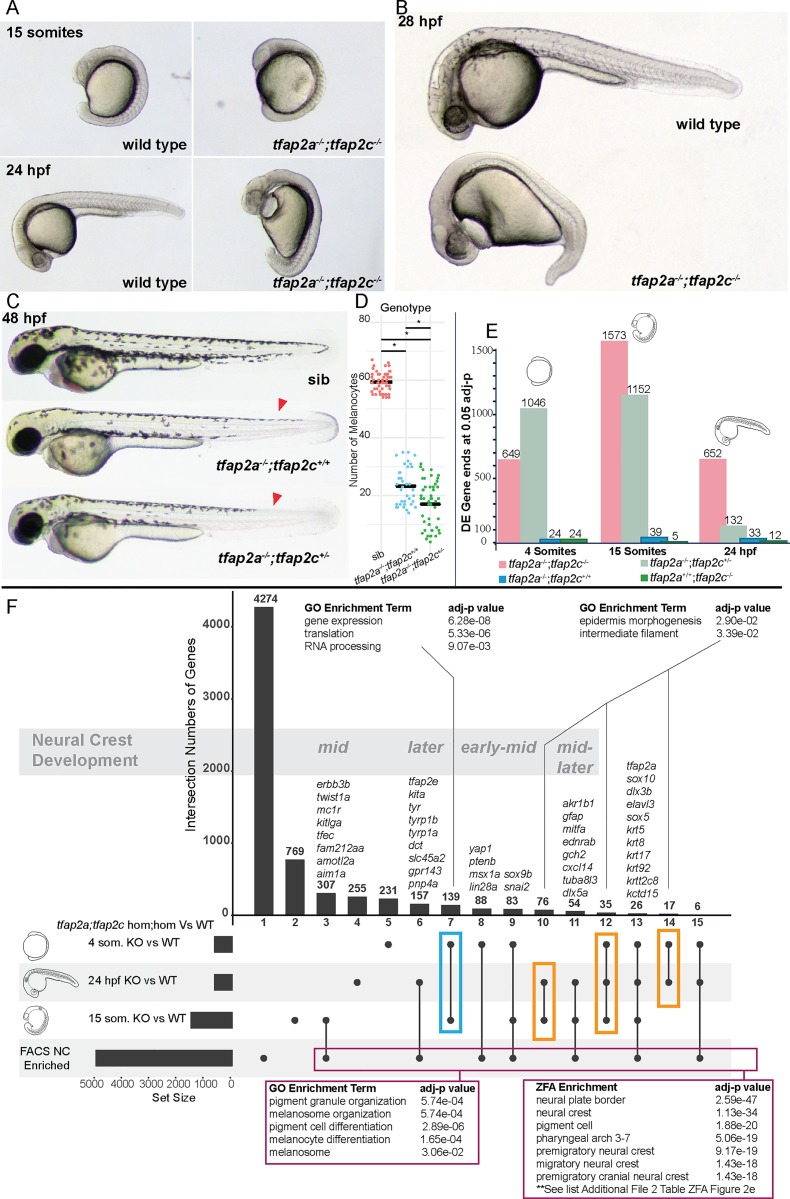
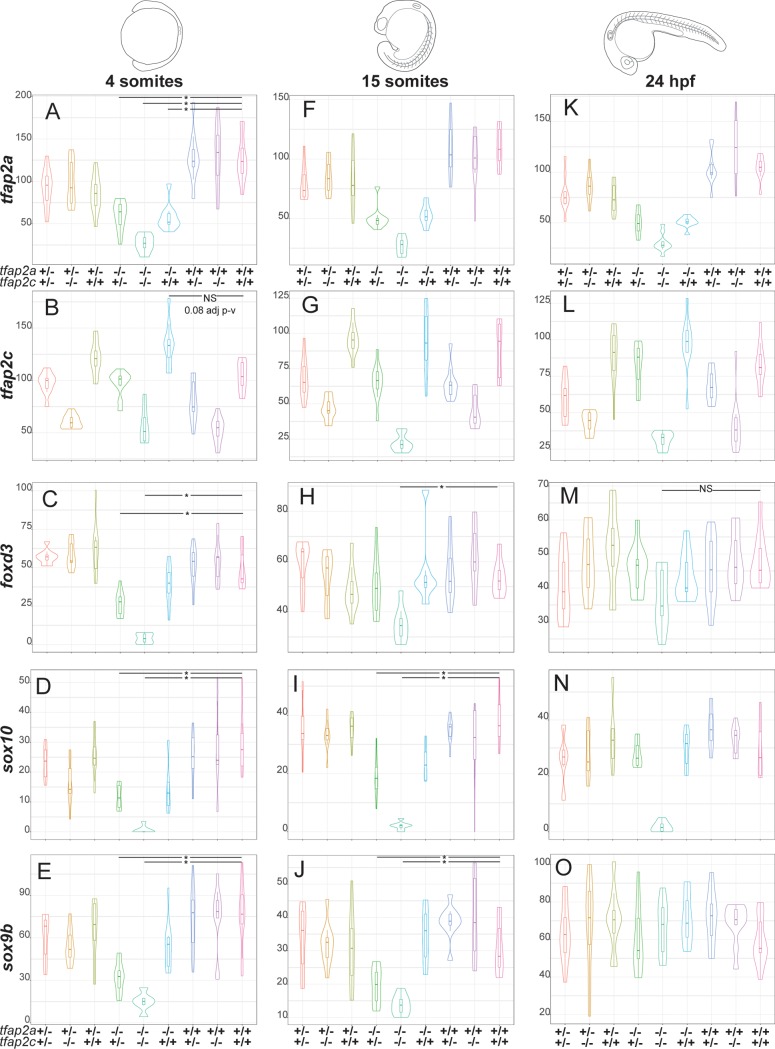
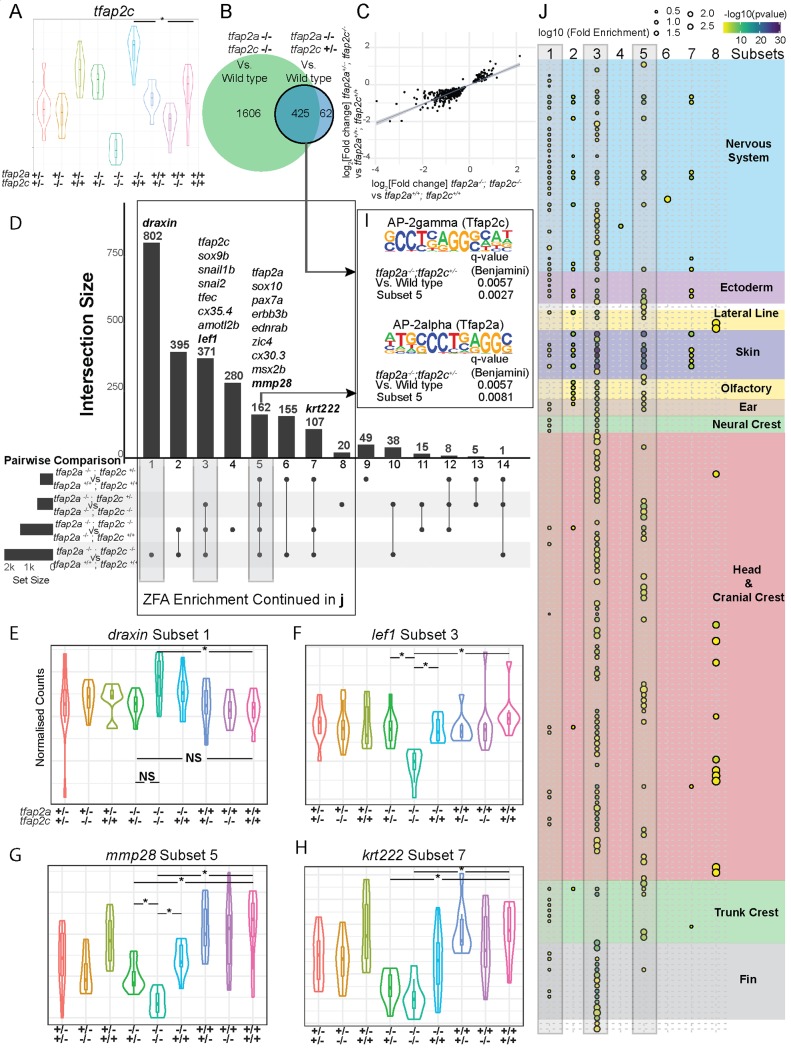
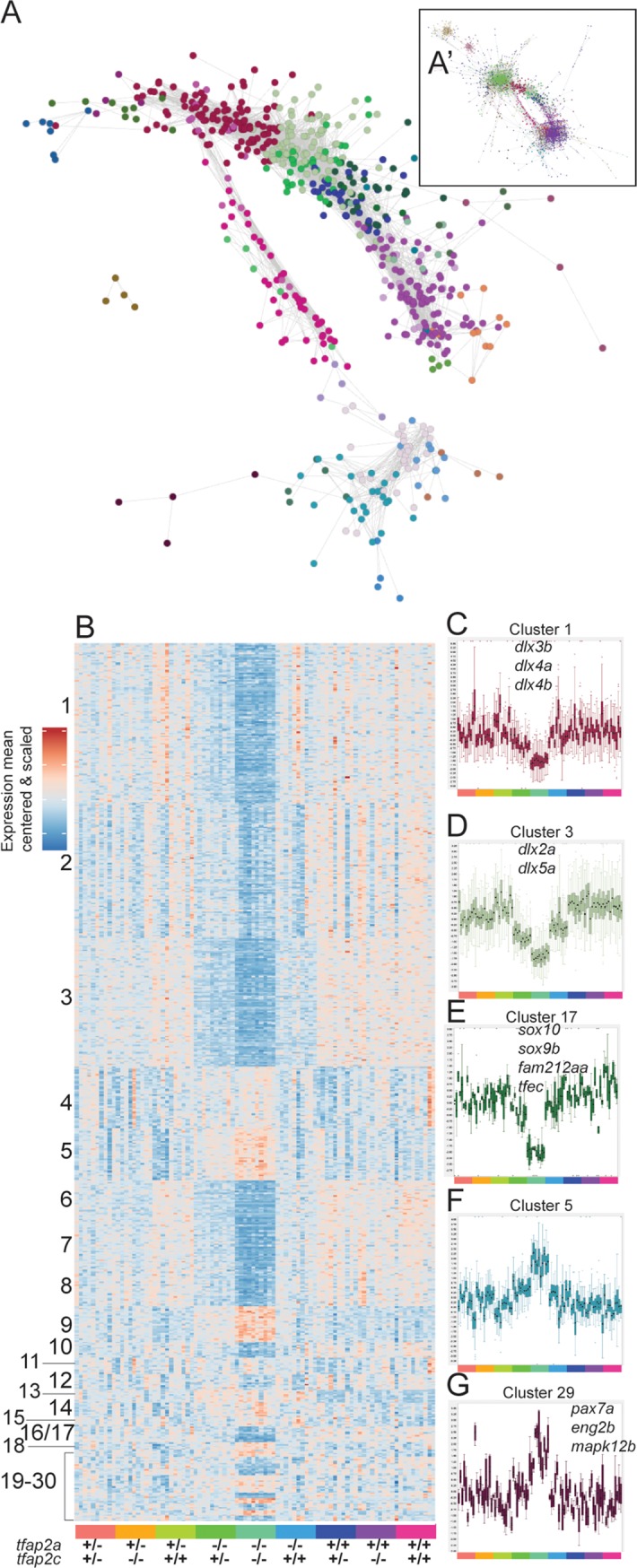
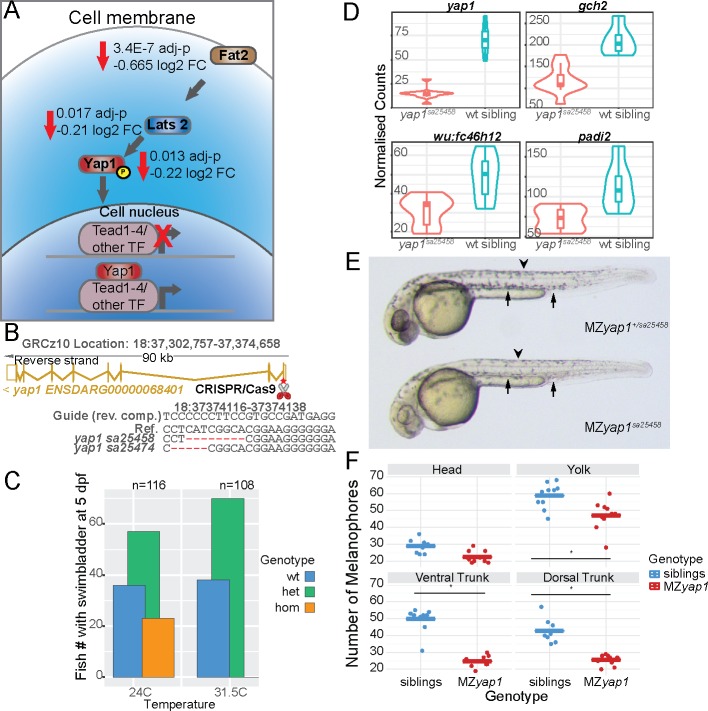
The neural crest (NC) is a vertebrate-specific cell type that contributes to a wide range of different tissues across all three germ layers. The gene regulatory network (GRN) responsible for the formation of neural crest is conserved across vertebrates. Central to the induction of the NC GRN are AP-2 and SoxE transcription factors. NC induction robustness is ensured through the ability of some of these transcription factors to compensate loss of function of gene family members. However the gene regulatory events underlying compensation are poorly understood. We have used gene knockout and RNA sequencing strategies to dissect NC induction and compensation in zebrafish. We genetically ablate the NC using double mutants of tfap2a;tfap2c or remove specific subsets of the NC with sox10 and mitfa knockouts and characterise genome-wide gene expression levels across multiple time points. We find that compensation through a single wild-type allele of tfap2c is capable of maintaining early NC induction and differentiation in the absence of tfap2a function, but many target genes have abnormal expression levels and therefore show sensitivity to the reduced tfap2 dosage. This separation of morphological and molecular phenotypes identifies a core set of genes required for early NC development. We also identify the 15 somites stage as the peak of the molecular phenotype which strongly diminishes at 24 hpf even as the morphological phenotype becomes more apparent. Using gene knockouts, we associate previously uncharacterised genes with pigment cell development and establish a role for maternal Hippo signalling in melanocyte differentiation. This work extends and refines the NC GRN while also uncovering the transcriptional basis of genetic compensation via paralogues.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Hall BK. The neural crest and neural crest cells in vertebrate development and evolution 2nd ed. New York: Springer; 2009.
-
- Le Douarin N, Kalcheim C. The neural crest 2. ed., digitally printed version. Cambridge: Cambridge Univ. Press; 2009.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
