

p65BTK is a novel potential actionable target in KRAS-mutated/EGFR-wild type lung adenocarcinoma
- PMID: 31200752
- PMCID: PMC6570906
- DOI: 10.1186/s13046-019-1199-7
p65BTK is a novel potential actionable target in KRAS-mutated/EGFR-wild type lung adenocarcinoma
Abstract
Background: Lung cancer is still the main cause of cancer death worldwide despite the availability of targeted therapies and immune-checkpoint inhibitors combined with chemotherapy. Cancer cell heterogeneity and primary or acquired resistance mechanisms cause the elusive behaviour of this cancer and new biomarkers and active drugs are urgently needed to overcome these limitations. p65BTK, a novel isoform of the Bruton Tyrosine Kinase may represent a new actionable target in non-small cell lung cancer (NSCLC).
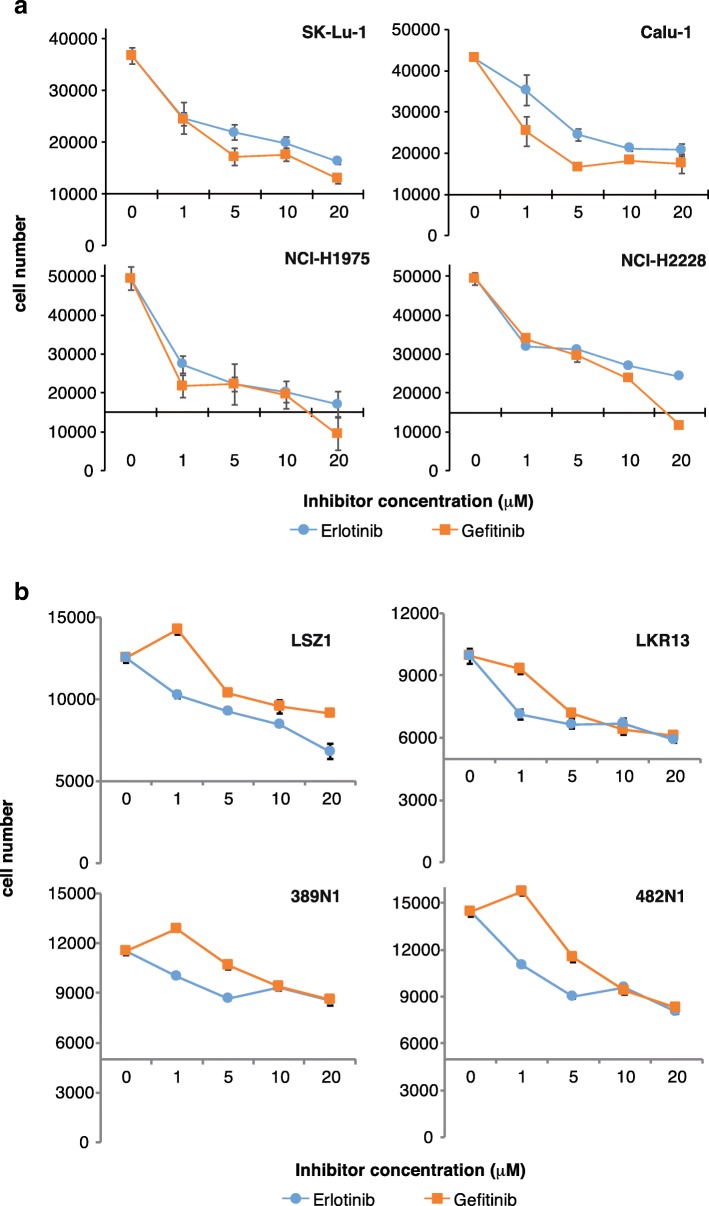
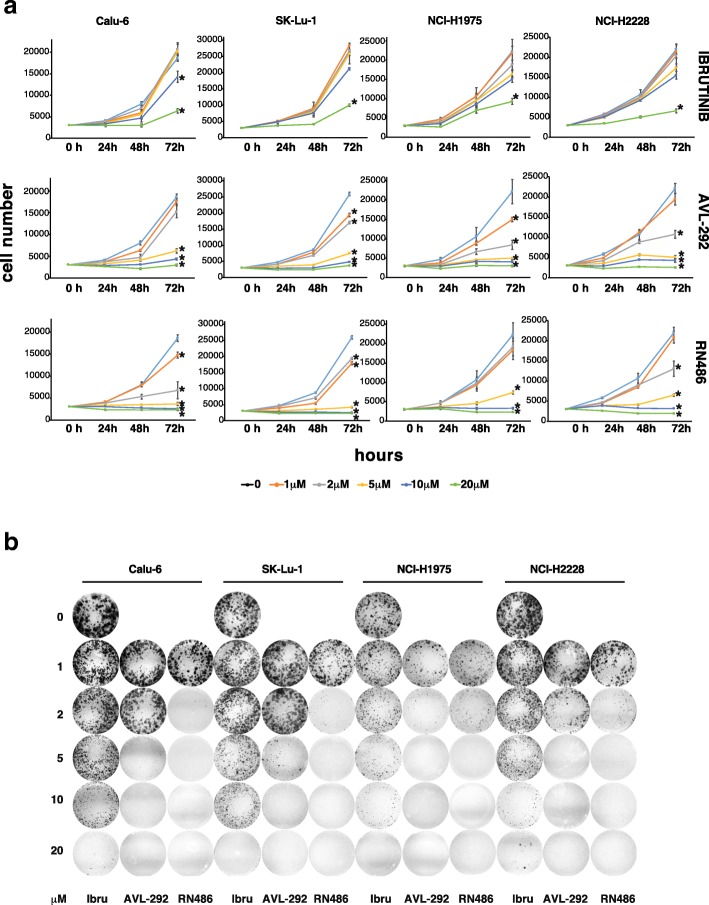
Methods: p65BTK expression was evaluated by immunohistochemistry in 382 NSCLC patients with complete clinico-pathological records including smoking habit, ALK and EGFR status, and in metastatic lymph nodes of 30 NSCLC patients. NSCLC cell lines mutated for p53 and/or a component of the RAS/MAPK pathway and primary lung cancer-derived cells from Kras/Trp53 null mice were used as a preclinical model. The effects of p65BTK inhibition by BTK Tyrosine Kinase Inhibitors (TKIs) (Ibrutinib, AVL-292, RN486) and first-generation EGFR-TKIs (Gefitinib, Erlotinib) on cell viability were evaluated by MTT. The effects of BTK-TKIs on cell growth and clonogenicity were assessed by crystal violet and colony assays, respectively. Cell toxicity assays were performed to study the effect of the combination of non-toxic concentrations of BTK-TKIs with EGFR-TKIs and standard-of-care (SOC) chemotherapy (Cisplatin, Gemcitabine, Pemetrexed).
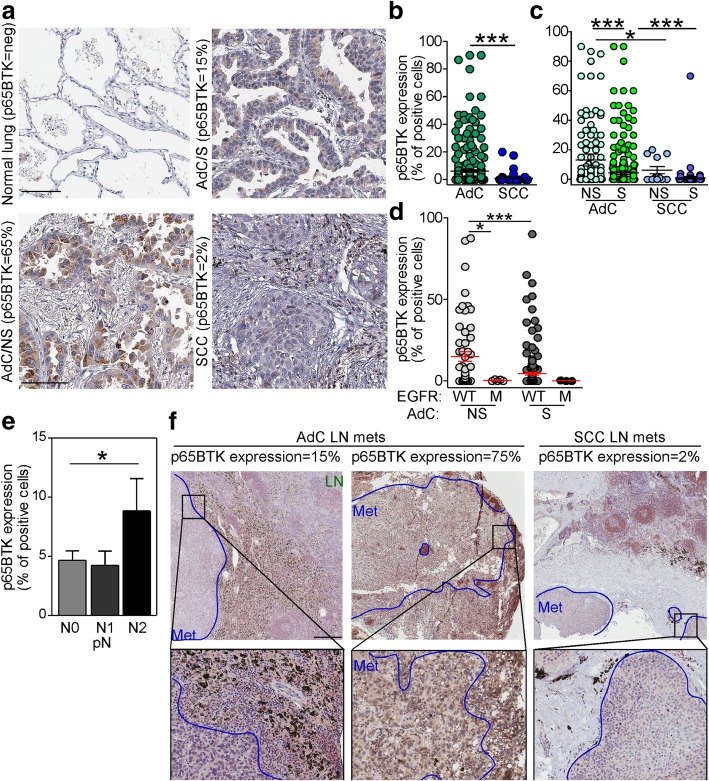
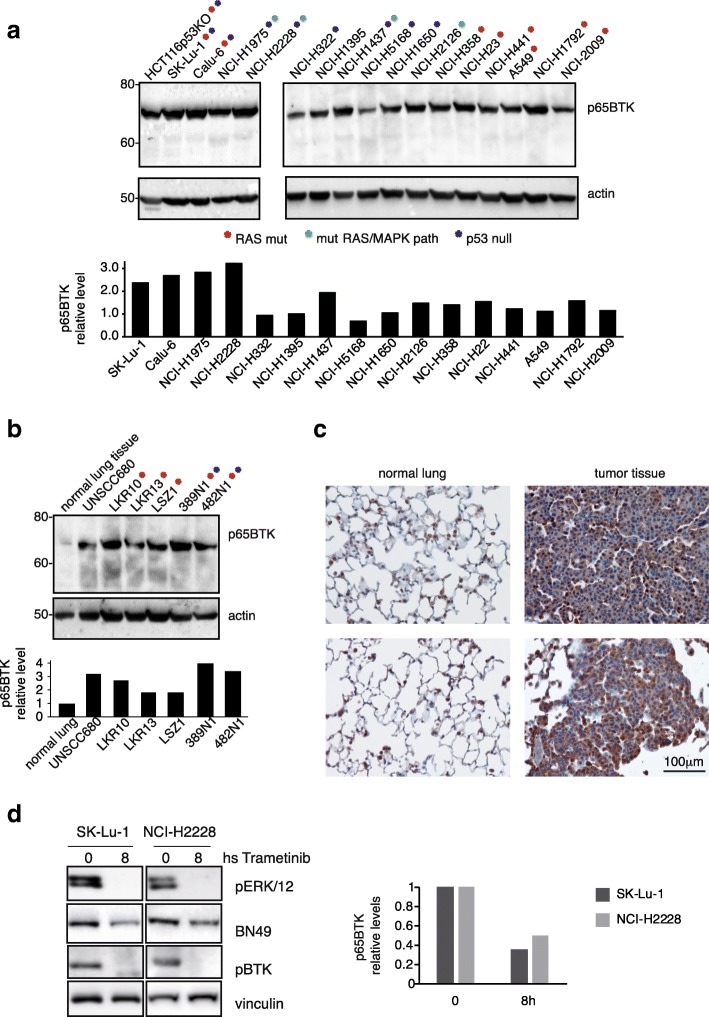
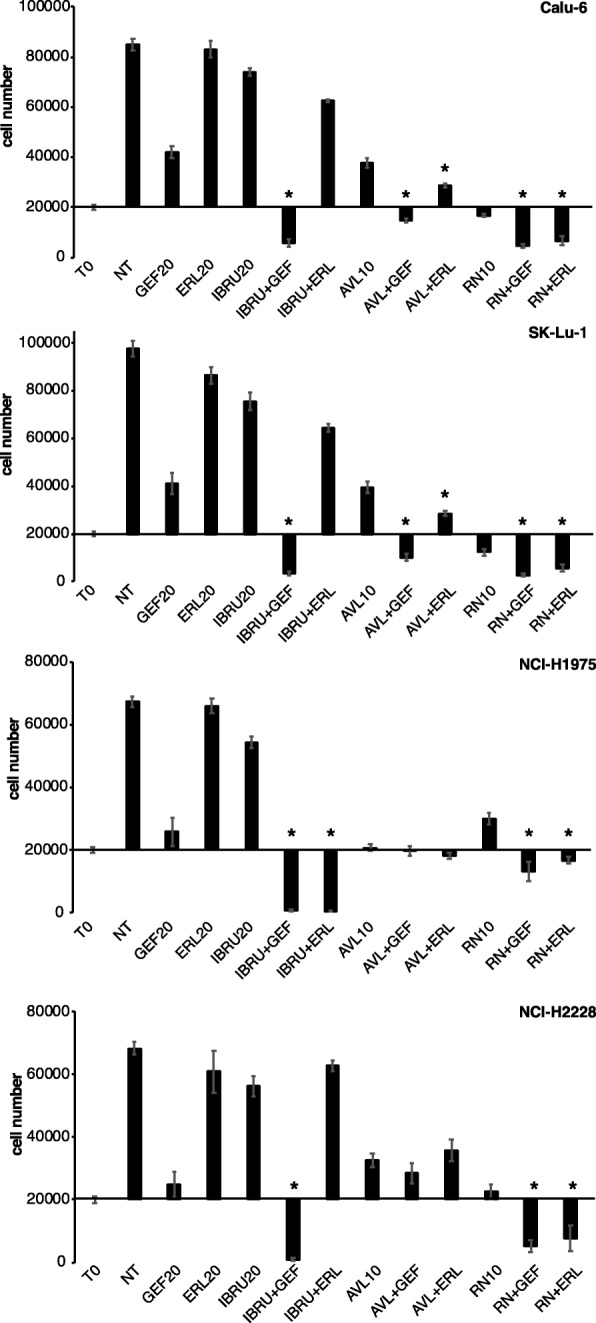
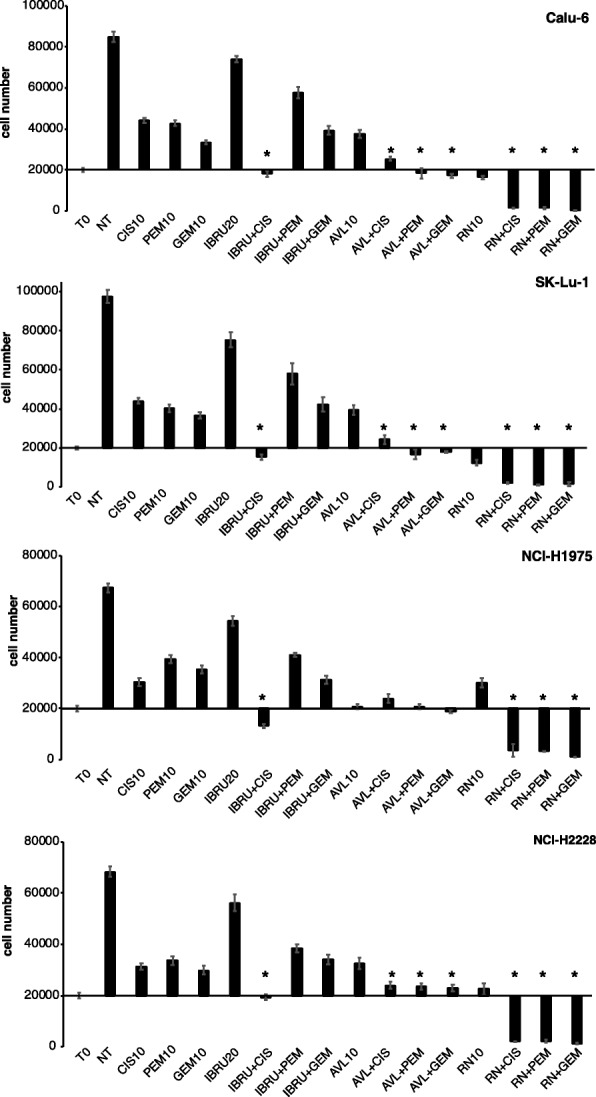
Results: p65BTK was significantly over-expressed in EGFR-wild type (wt) adenocarcinomas (AdC) from non-smoker patients and its expression was also preserved at the metastatic site. p65BTK was also over-expressed in cell lines mutated for KRAS or for a component of the RAS/MAPK pathway and in tumors from Kras/Trp53 null mice. BTK-TKIs were more effective than EGFR-TKIs in decreasing cancer cell viability and significantly impaired cell proliferation and clonogenicity. Moreover, non-toxic doses of BTK-TKIs re-sensitized drug-resistant NSCLC cell lines to both target- and SOC therapy, independently from EGFR/KRAS status.
Conclusions: p65BTK results as an emerging actionable target in non-smoking EGFR-wt AdC, also at advanced stages of disease. Notably, these patients are not eligible for EGFR-TKIs-based therapy due to a lack of EGFR mutation. The combination of BTK-TKIs with EGFR-TKIs is cytotoxic for EGFR-wt/KRAS-mutant/p53-null tumors and BTK-TKIs re-sensitizes drug-resistant NSCLC to SOC chemotherapy. Therefore, our data suggest that adding BTK-TKIs to SOC chemotherapy and EGFR-targeted therapy may open new avenues for clinical trials in currently untreatable NSCLC.
Keywords: BTK inhibitors; Chemotherapy; Drug resistance; EGFR; EGFR inhibitors; NSCLC; Targeted therapy; p65BTK.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Ye Q, She Q-B. Integration of AKT and ERK signaling pathways in Cancer: biological and therapeutic implications. J Pharmacol Clin Toxicol. 2013;1(2):1009.
MeSH terms
Substances
Grants and funding
- PON01_02782/Ministero dell'Istruzione, dell'Università e della Ricerca
- FAR 2016-ATE-0599/Università degli Studi di Milano-Bicocca
- Targeting p65BTK in Non-Small Cell Lung Cancer/Lung Cancer Research Foundation
- GR-2011-02351626/Ministero della Salute
- SAF2013-46423-R and SAF2017-89944-R/Ministerio de Economía y Competitividad
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
