

The p52 isoform of SHC1 is a key driver of breast cancer initiation
- PMID: 31202267
- PMCID: PMC6570928
- DOI: 10.1186/s13058-019-1155-7
The p52 isoform of SHC1 is a key driver of breast cancer initiation
Abstract
Background: SHC1 proteins (also called SHCA) exist in three functionally distinct isoforms (p46SHC, p52SHC, and p66SHC) that serve as intracellular adaptors for several key signaling pathways in breast cancer. Despite the broad evidence implicating SHC1 gene products as a central mediator of breast cancer, testing the isoform-specific roles of SHC1 proteins have been inaccessible due to the lack of isoform-specific inhibitors or gene knockout models.
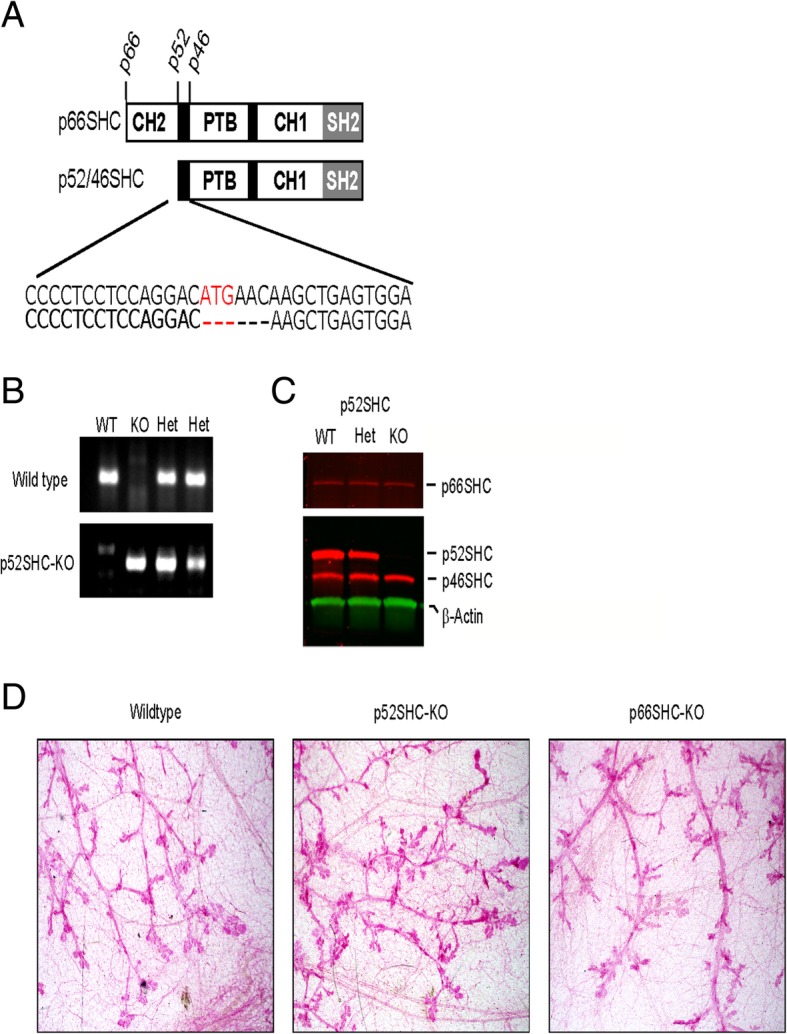
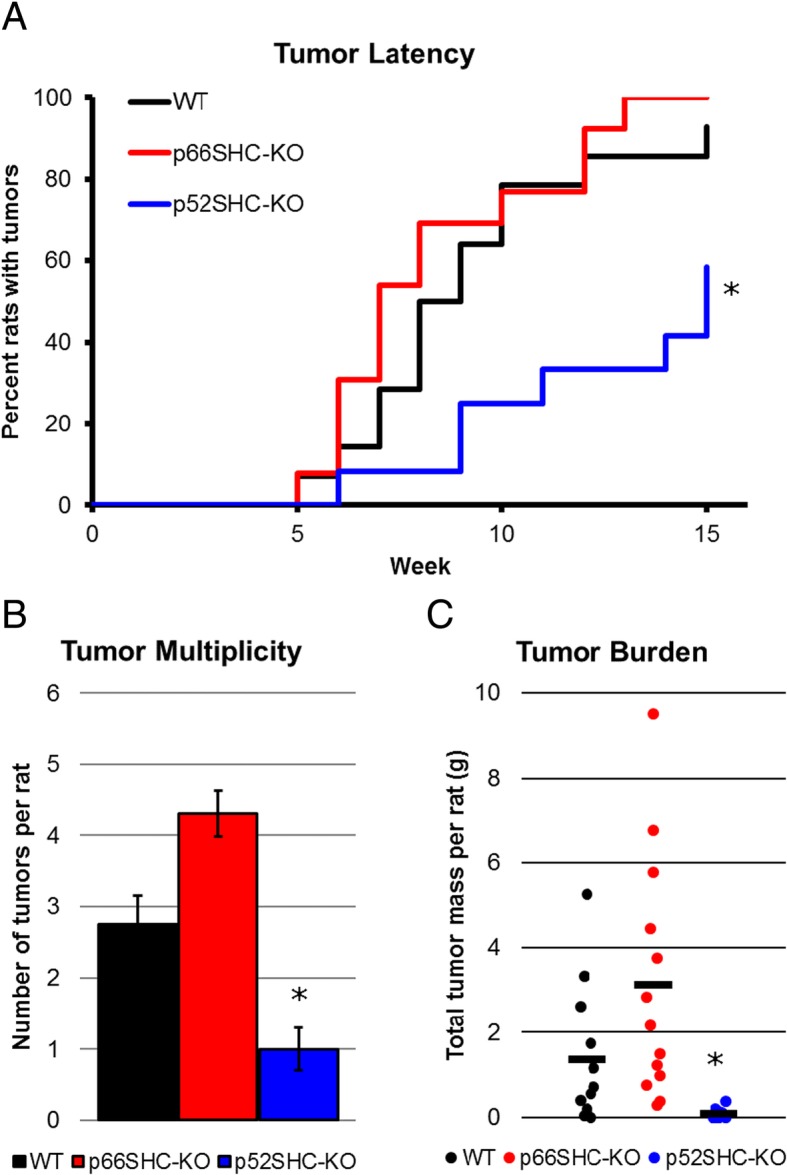
Methods: Here, we addressed this issue by generating the first isoform-specific gene knockout models for p52SHC and p66SHC, using germline gene editing in the salt-sensitive rat strain. Compared with the wild-type (WT) rats, we found that genetic ablation of the p52SHC isoform significantly attenuated mammary tumor formation, whereas the p66SHC knockout had no effect. Rats were dosed with 7,12-dimethylbenz(a)anthracene (DMBA) by oral gavage to induce mammary tumors, and progression of tumor development was followed for 15 weeks. At 15 weeks, tumors were excised and analyzed by RNA-seq to determine differences between tumors lacking p66SHC or p52SHC.
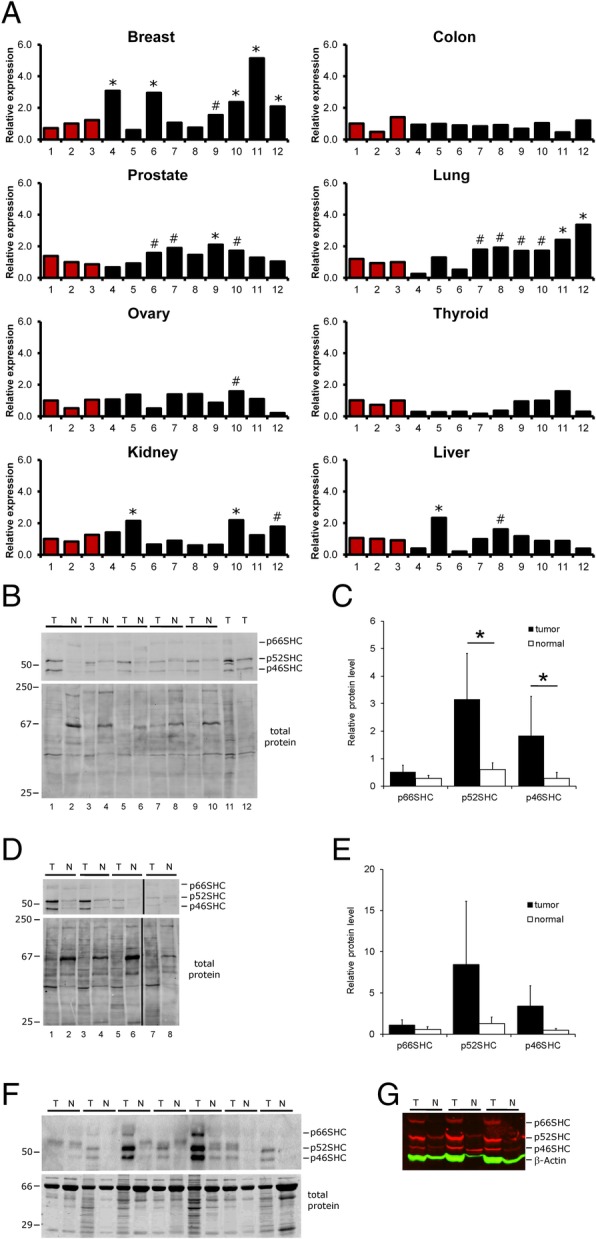
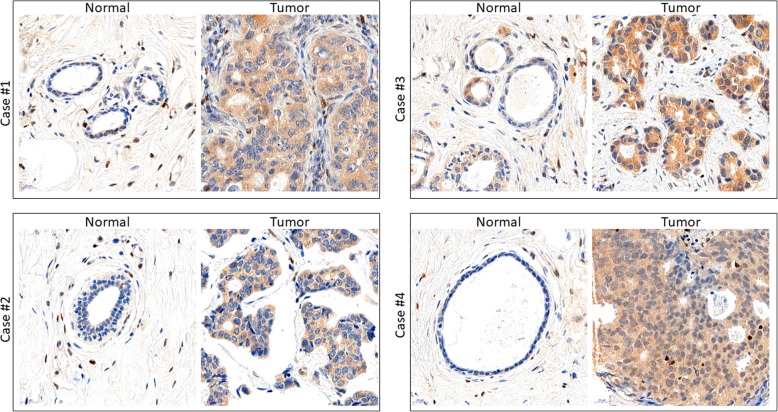
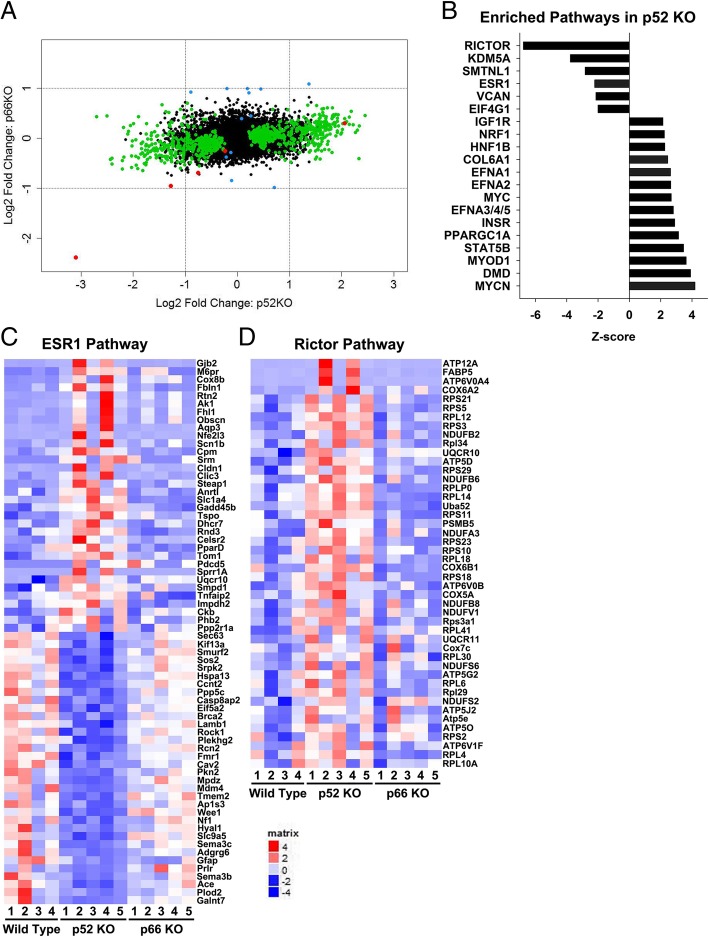
Results: Compared with the wild-type (WT) rats, we found that genetic ablation of the p52SHC isoform significantly attenuated mammary tumor formation, whereas the p66SHC knockout had no effect. These data, combined with p52SHC being the predominant isoform that is upregulated in human and rat tumors, provide the first evidence that p52SHC is the oncogenic isoform of Shc1 gene products in breast cancer. Compared with WT tumors, 893 differentially expressed (DE; FDR < 0.05) genes were detected in p52SHC KO tumors compared with only 18 DE genes in the p66SHC KO tumors, further highlighting that p52SHC is the relevant SHC1 isoform in breast cancer. Finally, gene network analysis revealed that p52SHC KO disrupted multiple key pathways that have been previously implicated in breast cancer initiation and progression, including ESR1 and mTORC2/RICTOR.
Conclusion: Collectively, these data demonstrate the p52SHC isoform is the key driver of DMBA-induced breast cancer while the expression of p66SHC and p46SHC are not enough to compensate.
Keywords: Breast cancer; DMBA; Rat model; Shc proteins; Signaling.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Das R, Vonderhaar BK. Involvement of SHC, GRB2, SOS and RAS in prolactin signal transduction in mammary epithelial cells. Oncogene. 1996;13(6):1139–1145. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
