

HumanMycobiomeScan: a new bioinformatics tool for the characterization of the fungal fraction in metagenomic samples
- PMID: 31202277
- PMCID: PMC6570844
- DOI: 10.1186/s12864-019-5883-y
HumanMycobiomeScan: a new bioinformatics tool for the characterization of the fungal fraction in metagenomic samples
Abstract
Background: Modern metagenomic analysis of complex microbial communities produces large amounts of sequence data containing information on the microbiome in terms of bacterial, archaeal, viral and eukaryotic composition. The bioinformatics tools available are mainly devoted to profiling the bacterial and viral fractions and only a few software packages consider fungi. As the human fungal microbiome (human mycobiome) can play an important role in the onset and progression of diseases, a comprehensive description of host-microbiota interactions cannot ignore this component.
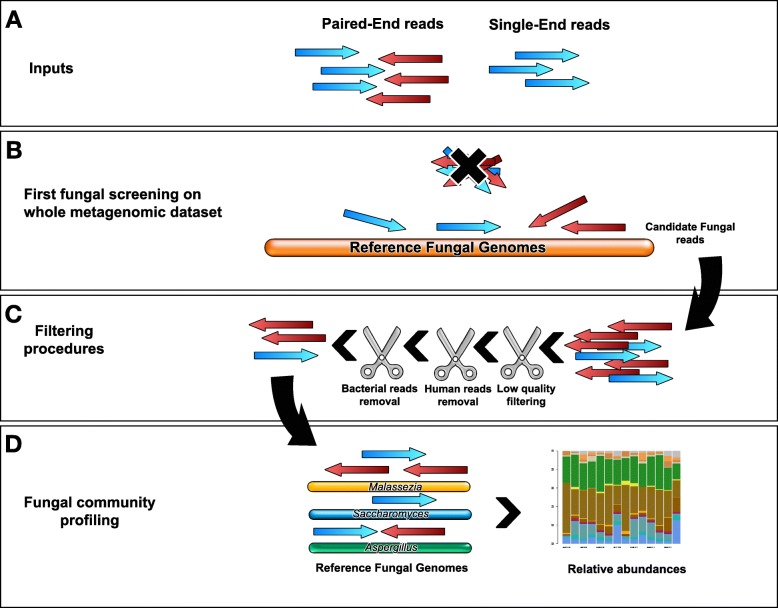
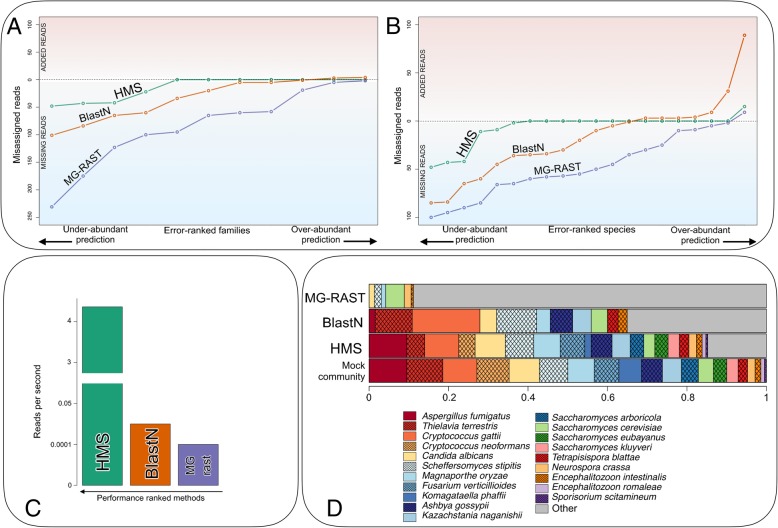
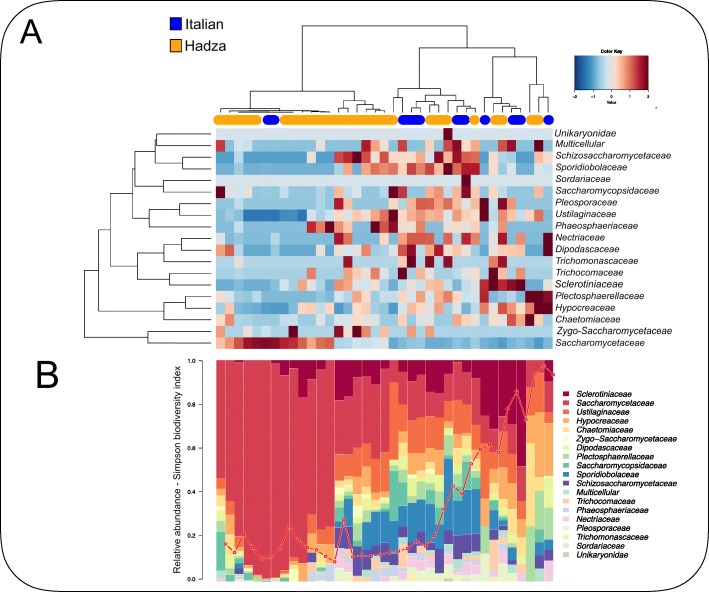
Results: HumanMycobiomeScan is a bioinformatics tool for the taxonomic profiling of the mycobiome directly from raw data of next-generation sequencing. The tool uses hierarchical databases of fungi in order to unambiguously assign reads to fungal species more accurately and > 10,000 times faster than other comparable approaches. HumanMycobiomeScan was validated using in silico generated synthetic communities and then applied to metagenomic data, to characterize the intestinal fungal components in subjects adhering to different subsistence strategies.
Conclusions: Although blind to unknown species, HumanMycobiomeScan allows the characterization of the fungal fraction of complex microbial ecosystems with good performance in terms of sample denoising from reads belonging to other microorganisms. HumanMycobiomeScan is most appropriate for well-studied microbiomes, for which most of the fungal species have been fully sequenced. This released version is functionally implemented to work with human-associated microbiota samples. In combination with other microbial profiling tools, HumanMycobiomeScan is a frugal and efficient tool for comprehensive characterization of microbial ecosystems through shotgun metagenomics sequencing.
Keywords: Metagenomics; Microbiome; Mycobiome.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
