

Glucosamine promotes hepatitis B virus replication through its dual effects in suppressing autophagic degradation and inhibiting MTORC1 signaling
- PMID: 31204557
- PMCID: PMC6999643
- DOI: 10.1080/15548627.2019.1632104
Glucosamine promotes hepatitis B virus replication through its dual effects in suppressing autophagic degradation and inhibiting MTORC1 signaling
Abstract
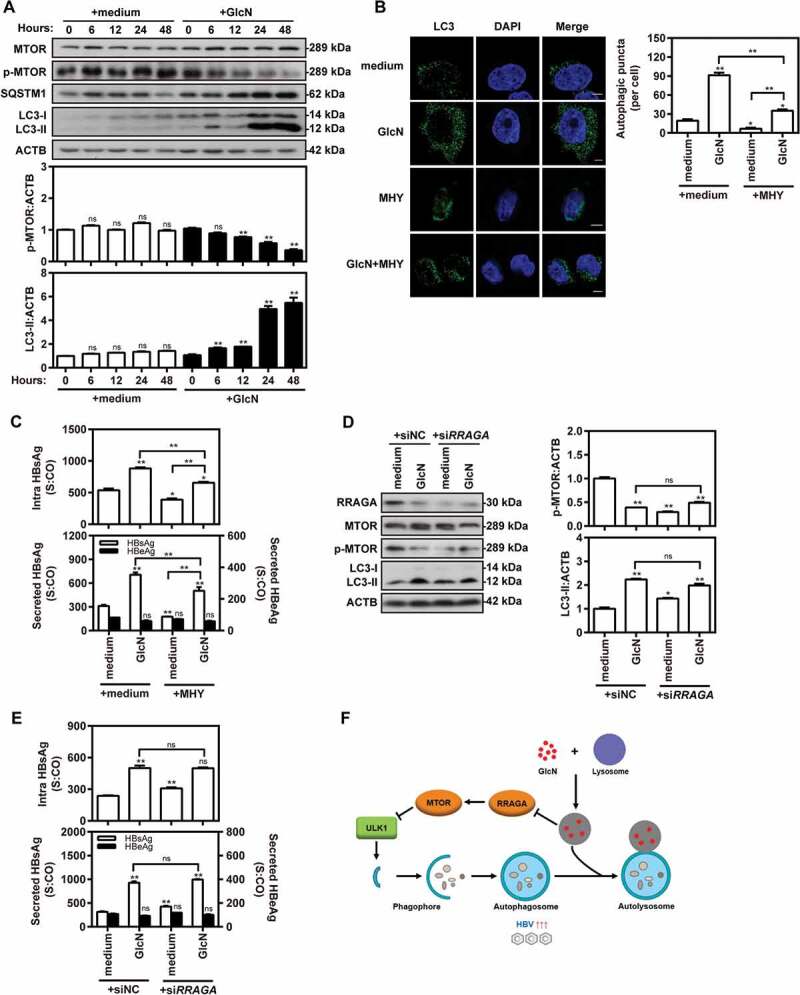
Glucosamine (GlcN), a dietary supplement widely utilized to promote joint health and effective in the treatment of osteoarthritis, is an effective macroautophagy/autophagy activator in vitro and in vivo. Previous studies have shown that autophagy is required for hepatitis B virus (HBV) replication and envelopment. The objective of this study was to determine whether and how GlcN affects HBV replication, using in vitro and in vivo experiments. Our data demonstrated that HBsAg production and HBV replication were significantly increased by GlcN treatment. Confocal microscopy and western blot analysis showed that the amount of autophagosomes and the levels of autophagic markers MAP1LC3/LC3-II and SQSTM1 were clearly elevated by GlcN treatment. GlcN strongly blocked autophagic degradation of HBV virions and proteins by inhibiting lysosomal acidification through its amino group. Moreover, GlcN further promoted HBV replication by inducing autophagosome formation via feedback inhibition of mechanistic target of rapamycin kinase complex 1 (MTORC1) signaling in an RRAGA (Ras related GTP binding A) GTPase-dependent manner. In vivo, GlcN application promoted HBV replication and blocked autophagic degradation in an HBV hydrodynamic injection mouse model. In addition, GlcN promoted influenza A virus, enterovirus 71, and vesicular stomatitis virus replication in vitro. In conclusion, GlcN efficiently promotes virus replication by inducing autophagic stress through its dual effects in suppressing autophagic degradation and inhibiting MTORC1 signaling. Thus, there is a potential risk of enhanced viral replication by oral GlcN intake in chronically virally infected patients.Abbreviations: ACTB: actin beta; ATG: autophagy-related; CMIA: chemiluminescence immunoassay; ConA: concanavalin A; CQ: chloroquine; CTSD: cathepsin D; DAPI: 4',6-diamidino-2-phenylindole; EV71: enterovirus 71; GalN: galactosamine; GFP: green fluorescence protein; GlcN: glucosamine; GNPNAT1: glucosamine-phosphate N-acetyltransferase 1; HBP: hexosamine biosynthesis pathway; HBV: hepatitis B virus; HBcAg: hepatitis B core antigen; HBsAg: hepatitis B surface antigen; HBeAg: hepatitis B e antigen; HBV RI: hepatitis B replicative intermediate; IAV: influenza A virus; LAMP1: lysosomal associated membrane protein 1; LAMTOR: late endosomal/lysosomal adaptor, MAPK and MTOR activator; ManN: mannosamine; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MTORC1: mechanistic target of rapamycin kinase complex 1; PHH: primary human hepatocyte; RAB7: RAB7A, member RAS oncogene family; RPS6KB1: ribosomal protein S6 kinase B1; RRAGA: Ras related GTP binding A; RT-PCR: reverse transcriptase polymerase chain reaction; SEM: standard error of the mean; siRNA: small interfering RNA; SQSTM1/p62: sequestosome 1; UAP1: UDP-N-acetylglucosamine pyrophosphorylase 1; VSV: vesicular stomatitis virus.
Keywords: Autophagy; HBV; MTORC1 signaling; glucosamine; lysosomal acidification.
Figures






Similar articles
-
CCDC88A/GIV promotes HBV replication and progeny secretion via enhancing endosomal trafficking and blocking autophagic degradation.Autophagy. 2022 Feb;18(2):357-374. doi: 10.1080/15548627.2021.1934271. Epub 2021 Jun 30. Autophagy. 2022. PMID: 34190023 Free PMC article.
-
Anti-hepatitis B virus activity of lithospermic acid, a polyphenol from Salvia miltiorrhiza, in vitro and in vivo by autophagy regulation.J Ethnopharmacol. 2023 Feb 10;302(Pt A):115896. doi: 10.1016/j.jep.2022.115896. Epub 2022 Nov 2. J Ethnopharmacol. 2023. PMID: 36334815
-
Hepatitis B virus promotes autophagic degradation but not replication in autophagosome.Biosci Trends. 2015 Apr;9(2):111-6. doi: 10.5582/bst.2015.01049. Biosci Trends. 2015. PMID: 25971696
-
Autophagy in farm animals: current knowledge and future challenges.Autophagy. 2021 Aug;17(8):1809-1827. doi: 10.1080/15548627.2020.1798064. Epub 2020 Jul 30. Autophagy. 2021. PMID: 32686564 Free PMC article. Review.
-
mTOR Signaling: The Interface Linking Cellular Metabolism and Hepatitis B Virus Replication.Virol Sin. 2021 Dec;36(6):1303-1314. doi: 10.1007/s12250-021-00450-3. Epub 2021 Sep 28. Virol Sin. 2021. PMID: 34580816 Free PMC article. Review.
Cited by
-
Characterization of SARS-CoV-2-specific humoral immunity and its potential applications and therapeutic prospects.Cell Mol Immunol. 2022 Feb;19(2):150-157. doi: 10.1038/s41423-021-00774-w. Epub 2021 Oct 13. Cell Mol Immunol. 2022. PMID: 34645940 Free PMC article. Review.
-
Autophagy in liver diseases.World J Hepatol. 2021 Jan 27;13(1):6-65. doi: 10.4254/wjh.v13.i1.6. World J Hepatol. 2021. PMID: 33584986 Free PMC article. Review.
-
The interplay between emerging human coronavirus infections and autophagy.Emerg Microbes Infect. 2021 Dec;10(1):196-205. doi: 10.1080/22221751.2021.1872353. Emerg Microbes Infect. 2021. PMID: 33399028 Free PMC article. Review.
-
mTOR Signaling: Roles in Hepatitis B Virus Infection and Hepatocellular Carcinoma.Int J Biol Sci. 2024 Aug 1;20(11):4178-4189. doi: 10.7150/ijbs.95894. eCollection 2024. Int J Biol Sci. 2024. PMID: 39247820 Free PMC article. Review.
-
Exosome-Autophagy Crosstalk in Enveloped Virus Infection.Int J Mol Sci. 2023 Jun 25;24(13):10618. doi: 10.3390/ijms241310618. Int J Mol Sci. 2023. PMID: 37445802 Free PMC article. Review.
References
-
- Skarbek K, Milewska MJ.. Biosynthetic and synthetic access to amino sugars. Carbohydr Res. 2016;434:44–71. - PubMed
-
- Reginster JY, Deroisy R, Rovati LC, et al. Long-term effects of glucosamine sulphate on osteoarthritis progression: a randomised, placebo-controlled clinical trial. Lancet. 2001;357:251–256. - PubMed
-
- Zahedipour F, Dalirfardouei R, Karimi G, et al. Molecular mechanisms of anticancer effects of Glucosamine. Biomed Pharmacother. 2017;95:1051–1058. - PubMed
-
- Liu X, Machado GC, Eyles JP, et al. Dietary supplements for treating osteoarthritis: a systematic review and meta-analysis. Br J Sports Med. 2018;52:167–175. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous