

TOX is a critical regulator of tumour-specific T cell differentiation
- PMID: 31207604
- PMCID: PMC7698992
- DOI: 10.1038/s41586-019-1324-y
TOX is a critical regulator of tumour-specific T cell differentiation
Abstract
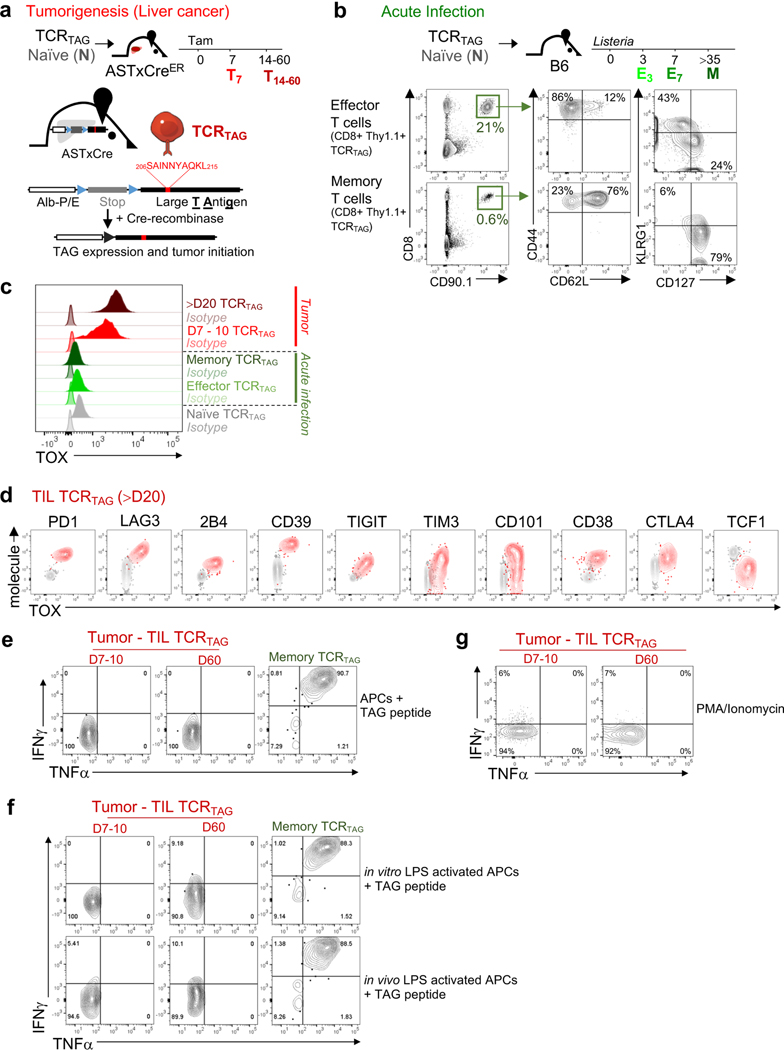
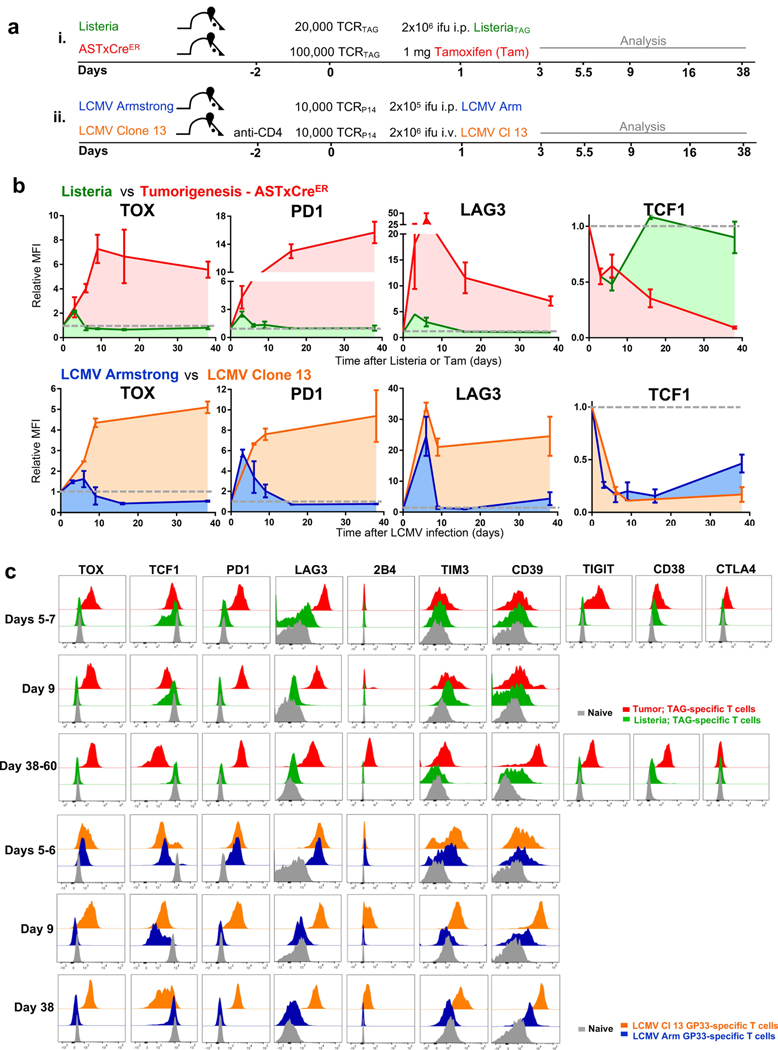
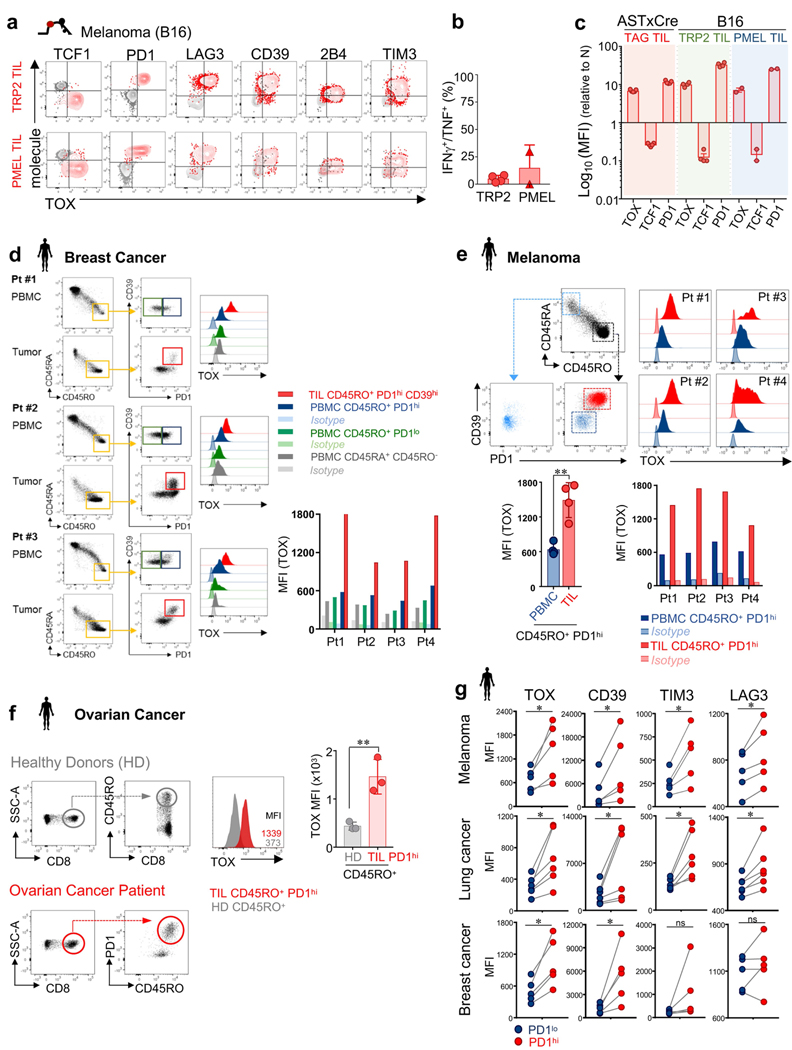
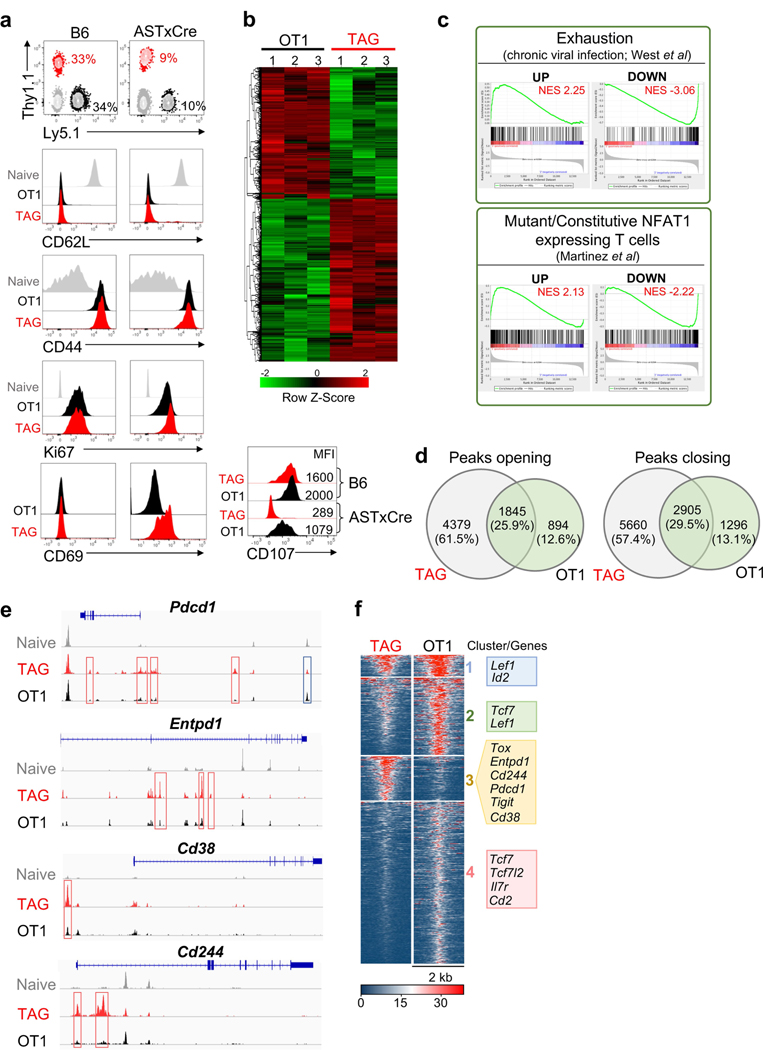
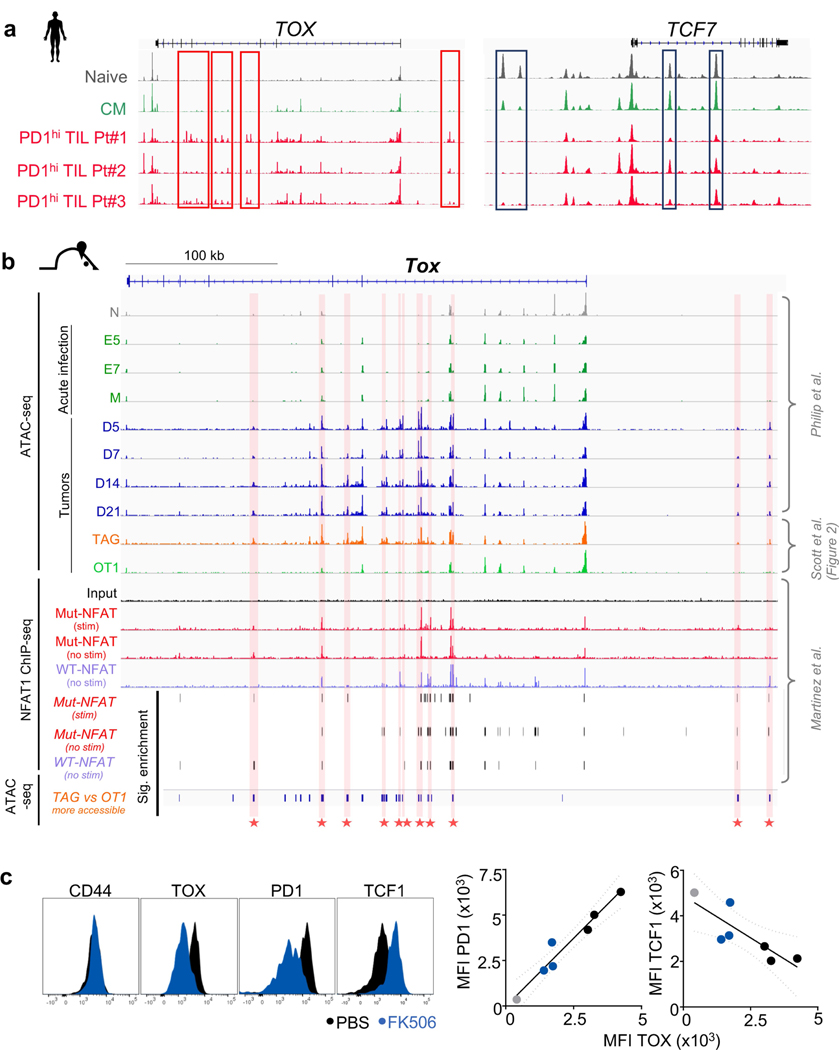
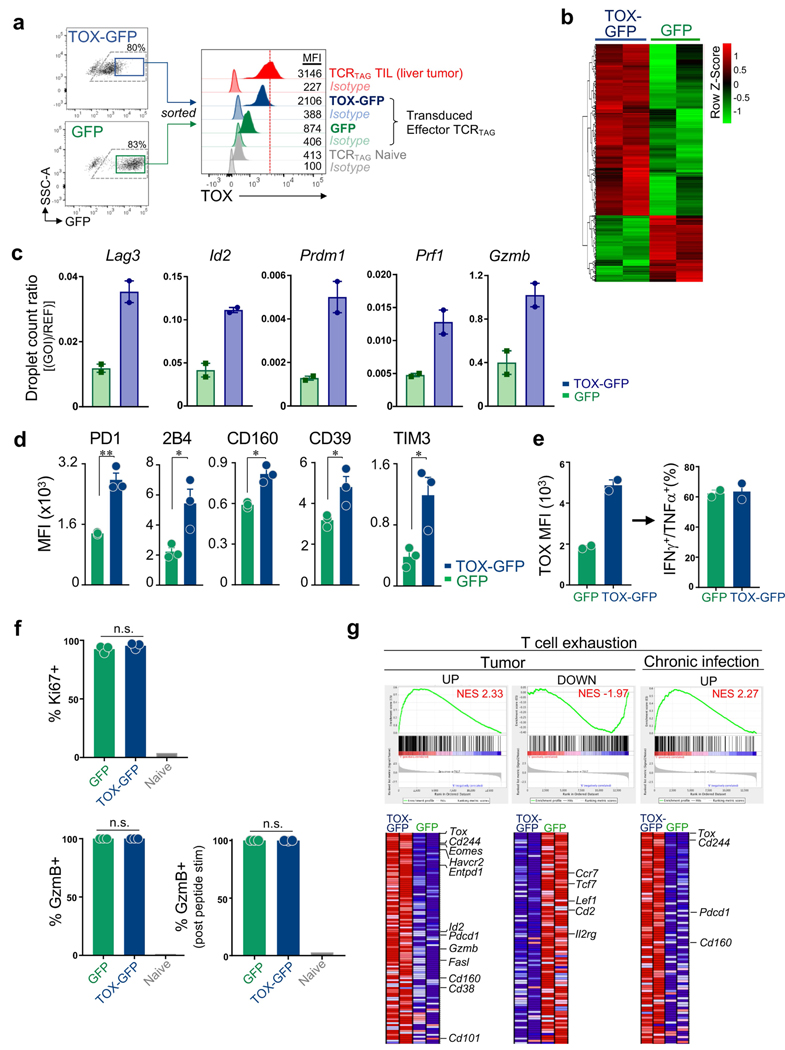
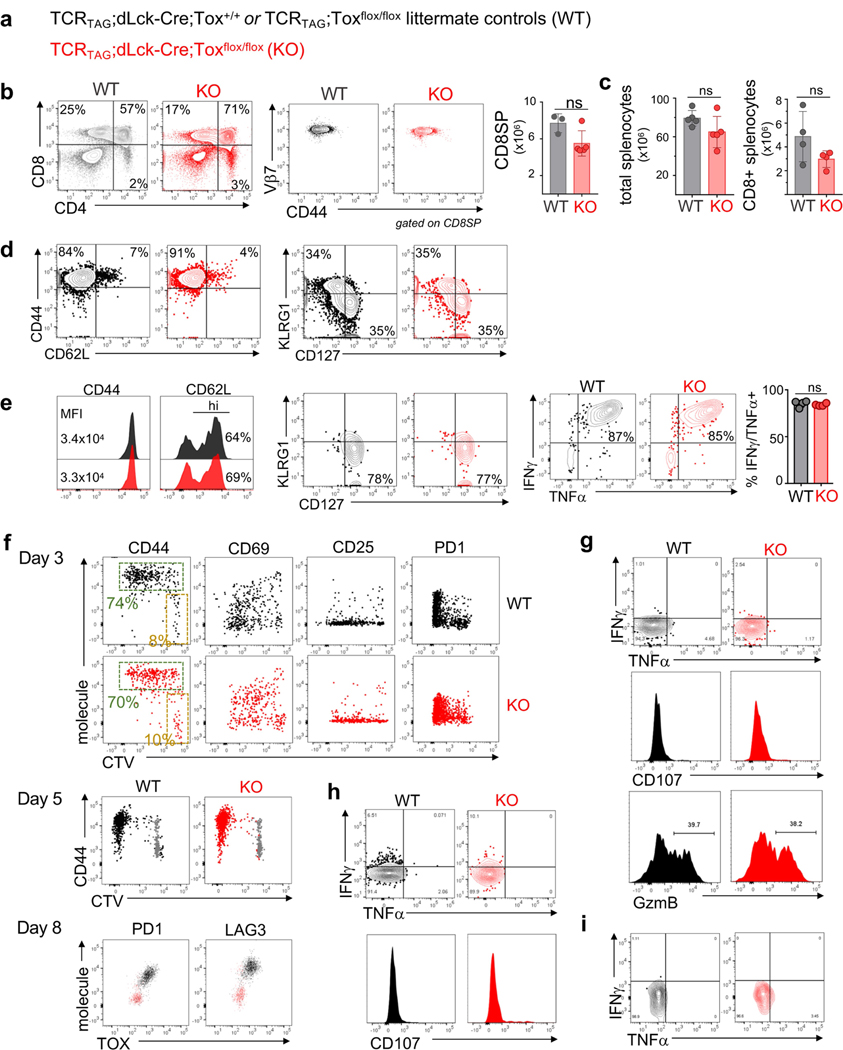
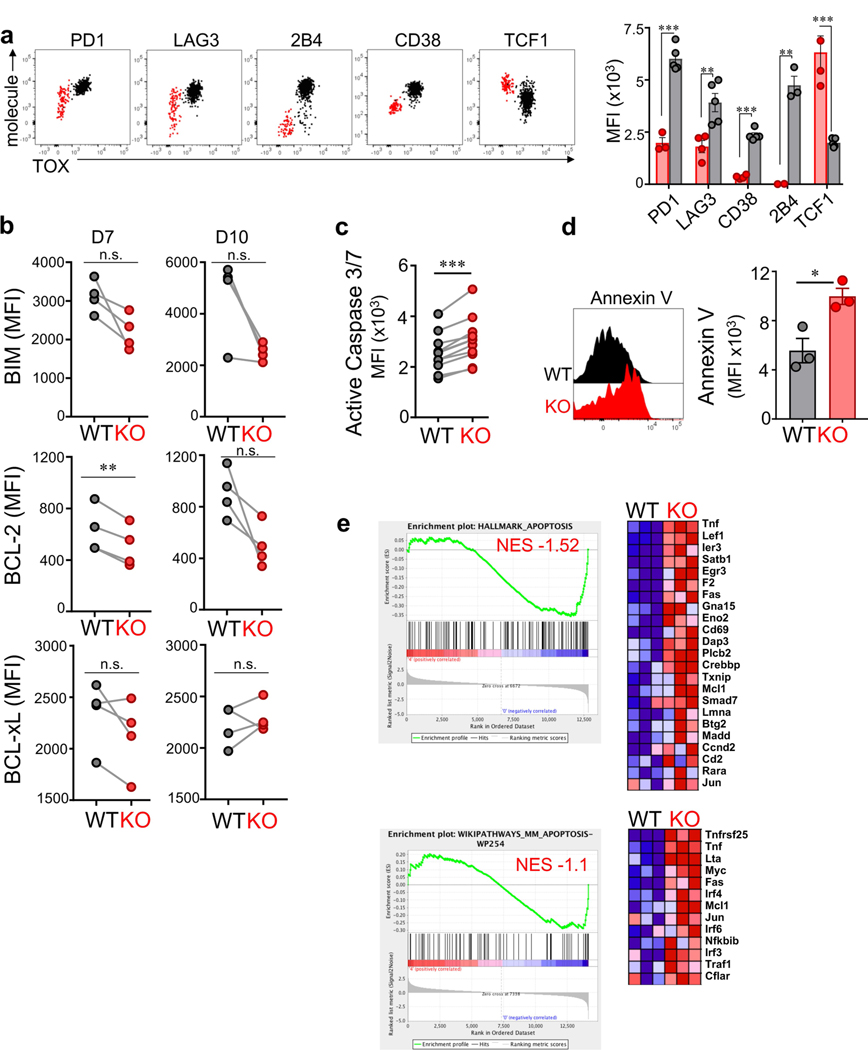
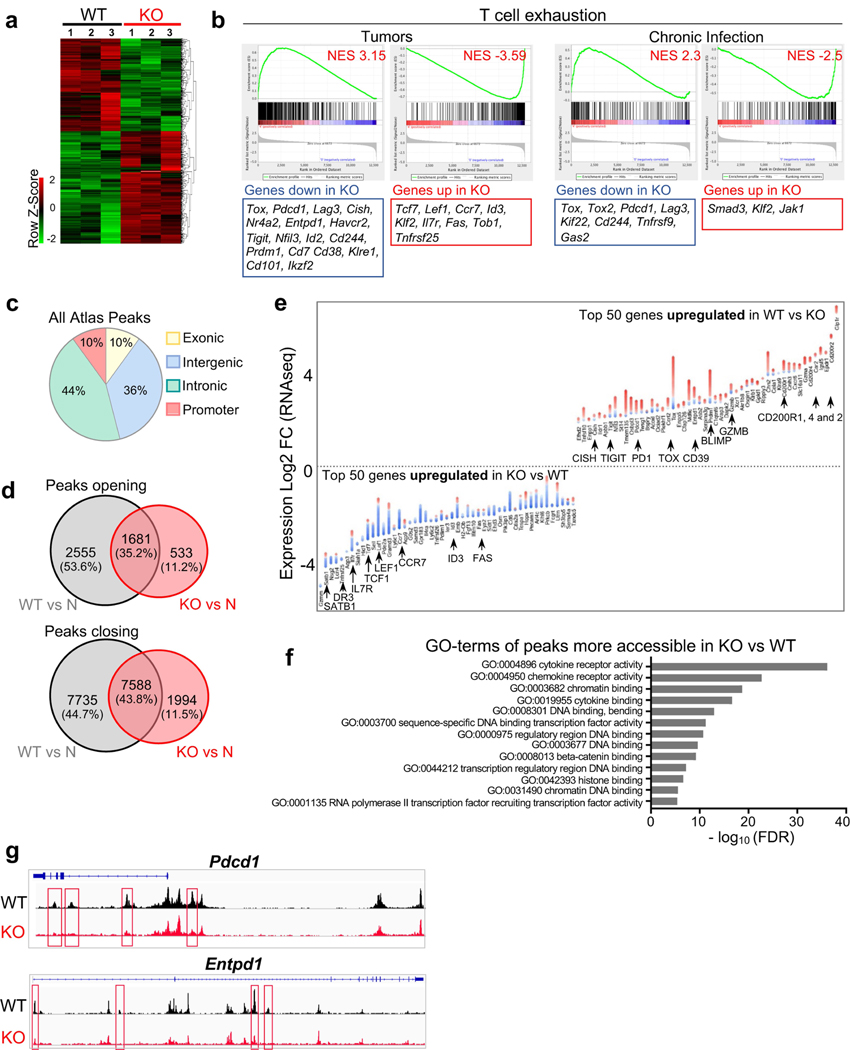
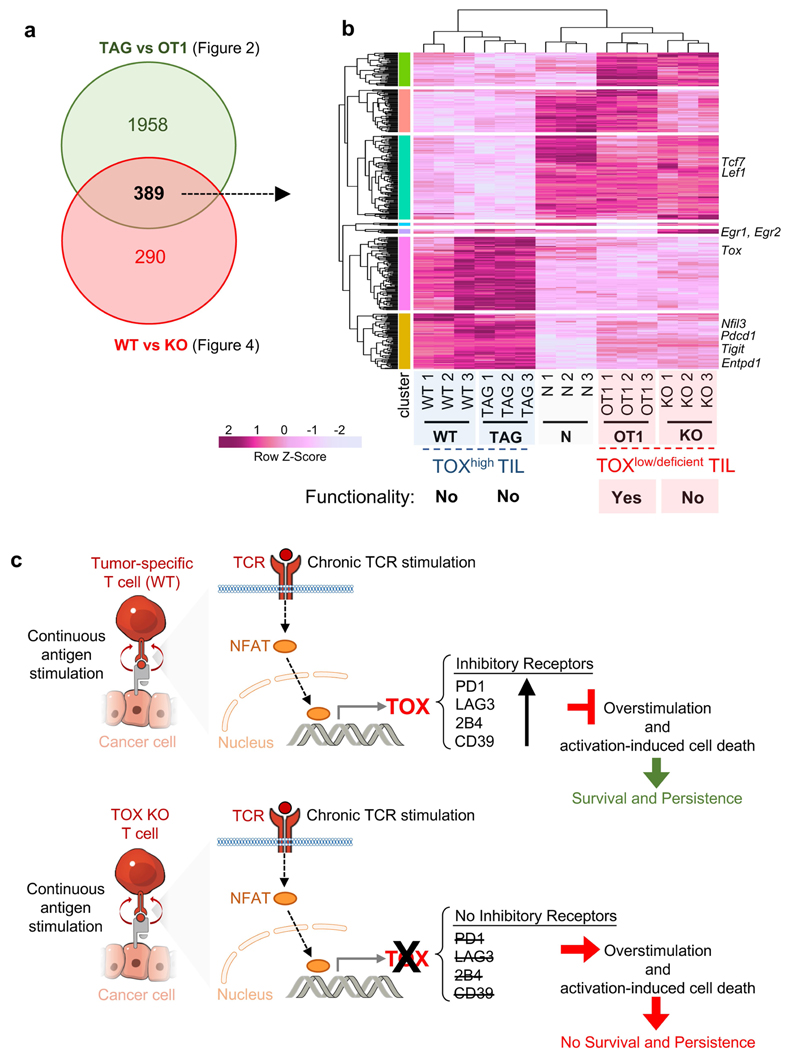
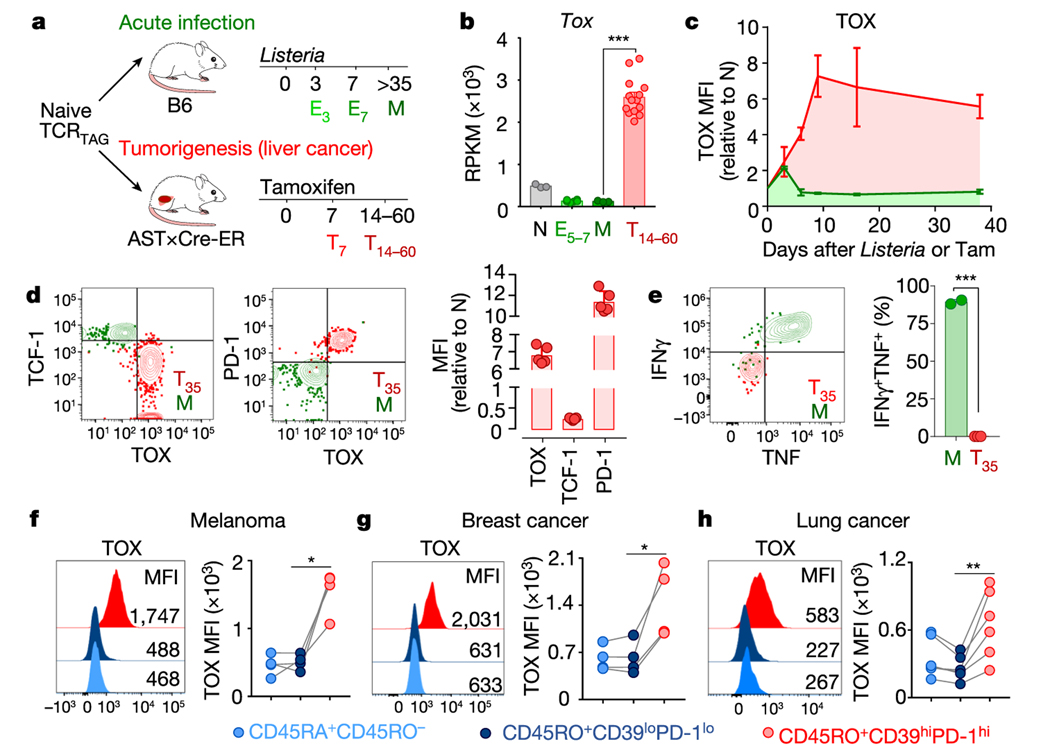
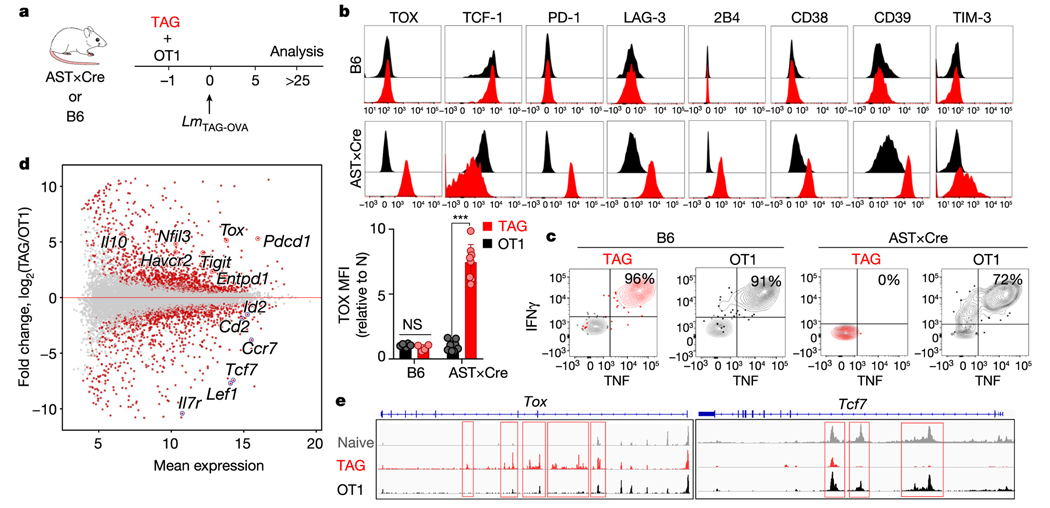
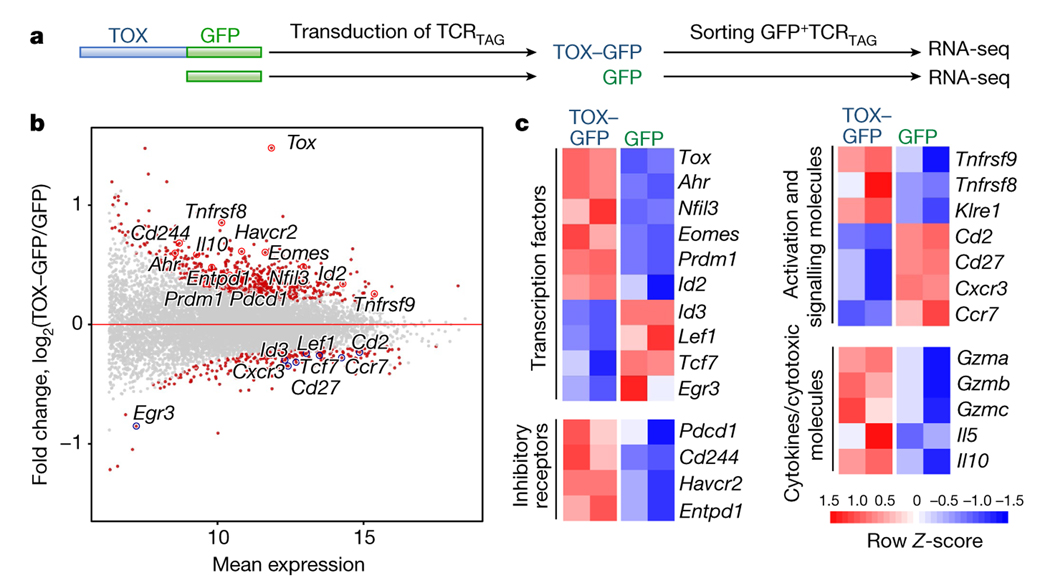
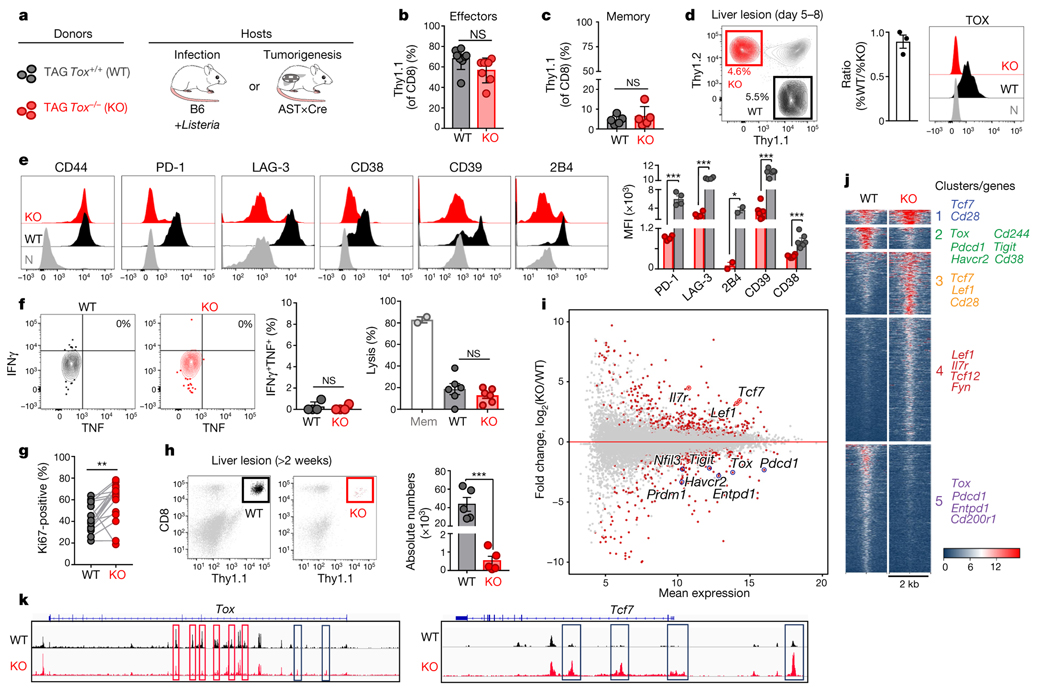
Tumour-specific CD8 T cell dysfunction is a differentiation state that is distinct from the functional effector or memory T cell states1-6. Here we identify the nuclear factor TOX as a crucial regulator of the differentiation of tumour-specific T (TST) cells. We show that TOX is highly expressed in dysfunctional TST cells from tumours and in exhausted T cells during chronic viral infection. Expression of TOX is driven by chronic T cell receptor stimulation and NFAT activation. Ectopic expression of TOX in effector T cells in vitro induced a transcriptional program associated with T cell exhaustion. Conversely, deletion of Tox in TST cells in tumours abrogated the exhaustion program: Tox-deleted TST cells did not upregulate genes for inhibitory receptors (such as Pdcd1, Entpd1, Havcr2, Cd244 and Tigit), the chromatin of which remained largely inaccessible, and retained high expression of transcription factors such as TCF-1. Despite their normal, 'non-exhausted' immunophenotype, Tox-deleted TST cells remained dysfunctional, which suggests that the regulation of expression of inhibitory receptors is uncoupled from the loss of effector function. Notably, although Tox-deleted CD8 T cells differentiated normally to effector and memory states in response to acute infection, Tox-deleted TST cells failed to persist in tumours. We hypothesize that the TOX-induced exhaustion program serves to prevent the overstimulation of T cells and activation-induced cell death in settings of chronic antigen stimulation such as cancer.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
