

Structural and Functional Impact of Seven Missense Variants of Phenylalanine Hydroxylase
- PMID: 31208052
- PMCID: PMC6628251
- DOI: 10.3390/genes10060459
Structural and Functional Impact of Seven Missense Variants of Phenylalanine Hydroxylase
Abstract
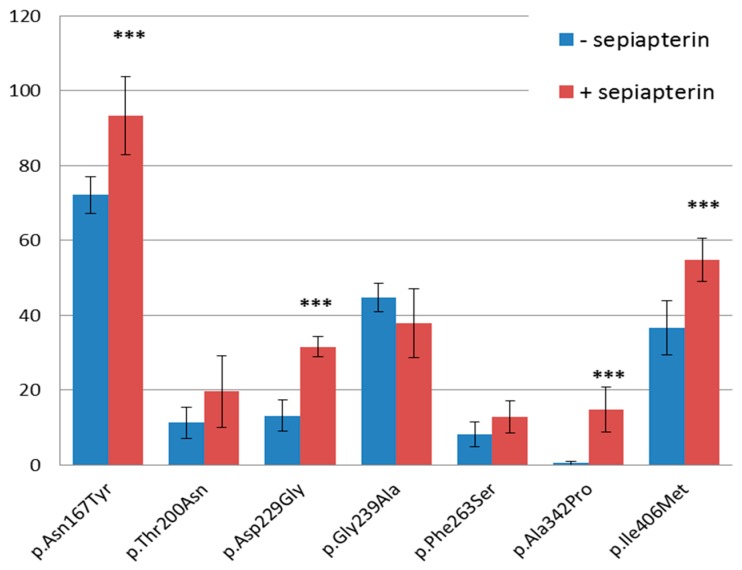
The molecular genetics of well-characterized inherited diseases, such as phenylketonuria (PKU) and hyperphenylalaninemia (HPA) predominantly caused by mutations in the phenylalanine hydroxylase (PAH) gene, is often complicated by the identification of many novel variants, often with no obvious impact on the associated disorder. To date, more than 1100 PAH variants have been identified of which a substantial portion have unknown clinical significance. In this work, we study the functionality of seven yet uncharacterized PAH missense variants p.Asn167Tyr, p.Thr200Asn, p.Asp229Gly, p.Gly239Ala, p.Phe263Ser, p.Ala342Pro, and p.Ile406Met first identified in the Czech PKU/HPA patients. From all tested variants, three of them, namely p.Asn167Tyr, p.Thr200Asn, and p.Ile406Met, exerted residual enzymatic activity in vitro similar to wild type (WT) PAH, however, when expressed in HepG2 cells, their protein level reached a maximum of 72.1% ± 4.9%, 11.2% ± 4.2%, and 36.6% ± 7.3% compared to WT PAH, respectively. Remaining variants were null with no enzyme activity and decreased protein levels in HepG2 cells. The chaperone-like effect of applied BH4 precursor increased protein level significantly for p.Asn167Tyr, p.Asp229Gly, p.Ala342Pro, and p.Ile406Met. Taken together, our results of functional characterization in combination with in silico prediction suggest that while p.Asn167Tyr, p.Thr200Asn, and p.Ile406Met PAH variants have a mild impact on the protein, p.Asp229Gly, p.Gly239Ala, p.Phe263Ser, and p.Ala342Pro severely affect protein structure and function.
Keywords: BH4; functional studies; missense variants; phenylalanine hydroxylase; phenylketonuria.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Hyperphenylalaninemia in the Czech Republic: genotype-phenotype correlations and in silico analysis of novel missense mutations.Clin Chim Acta. 2013 Apr 18;419:1-10. doi: 10.1016/j.cca.2013.01.006. Epub 2013 Jan 26. Clin Chim Acta. 2013. PMID: 23357515
-
Functional and structural characterisation of 5 missense mutations of the phenylalanine hydroxylase.Gen Physiol Biophys. 2017 Oct;36(4):361-371. doi: 10.4149/gpb_2017003. Epub 2017 Jun 27. Gen Physiol Biophys. 2017. PMID: 28653649
-
New protein structures provide an updated understanding of phenylketonuria.Mol Genet Metab. 2017 Aug;121(4):289-296. doi: 10.1016/j.ymgme.2017.06.005. Epub 2017 Jun 15. Mol Genet Metab. 2017. PMID: 28645531 Free PMC article. Review.
-
Functional Characterization of Novel Phenylalanine Hydroxylase p.Gln226Lys Mutation Revealed Its Non-responsiveness to Tetrahydrobiopterin Treatment in Hepatoma Cellular Model.Biochem Genet. 2018 Oct;56(5):533-541. doi: 10.1007/s10528-018-9858-5. Epub 2018 Apr 13. Biochem Genet. 2018. PMID: 29654578
-
Structural studies on phenylalanine hydroxylase and implications toward understanding and treating phenylketonuria.Pediatrics. 2003 Dec;112(6 Pt 2):1557-65. Pediatrics. 2003. PMID: 14654665 Review.
Cited by
-
Protein Degradation and the Pathologic Basis of Phenylketonuria and Hereditary Tyrosinemia.Int J Mol Sci. 2020 Jul 15;21(14):4996. doi: 10.3390/ijms21144996. Int J Mol Sci. 2020. PMID: 32679806 Free PMC article. Review.
References
-
- Lysinová M., Knapková M., Dluholucký S., Králinský K. Novorodenecký skríning v súčasnosti. Pediatr. Prax. 2015;16:188–191.
-
- Neonatal Screening in the Czech Republic. [(accessed on 3 April 2019)]; Available online: https://www.novorozeneckyscreening.cz/en.
-
- Guldberg P., Rey F., Zschocke J., Romano V., Baudouin F., Michiels L., Ullrich K., Hoffmann G.F., Burgard P., Schmidt H., et al. A European multicenter study of phenylalanine hydroxylase deficiency: Classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am. J. Hum. Genet. 1998;63:71–79. doi: 10.1086/301920. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
