

Quantitative evidence for early metastatic seeding in colorectal cancer
- PMID: 31209394
- PMCID: PMC6982526
- DOI: 10.1038/s41588-019-0423-x
Quantitative evidence for early metastatic seeding in colorectal cancer
Abstract
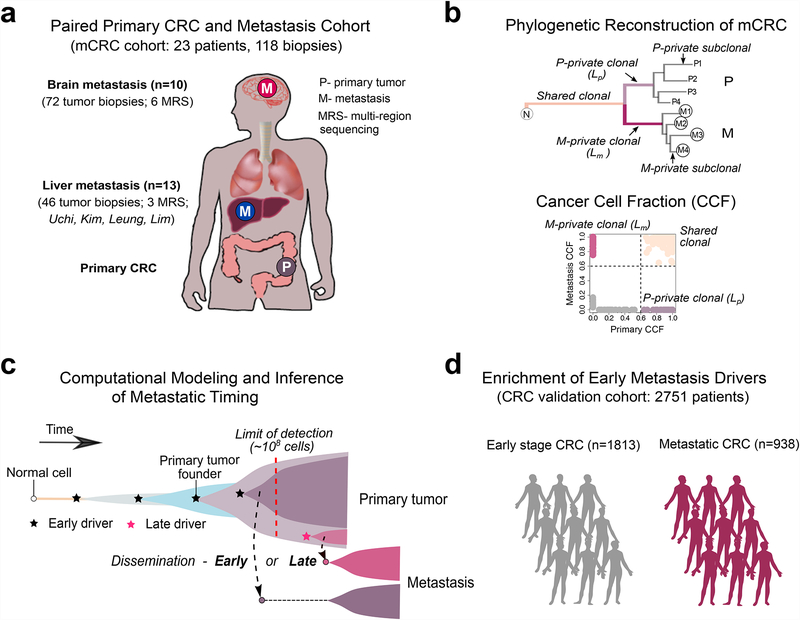
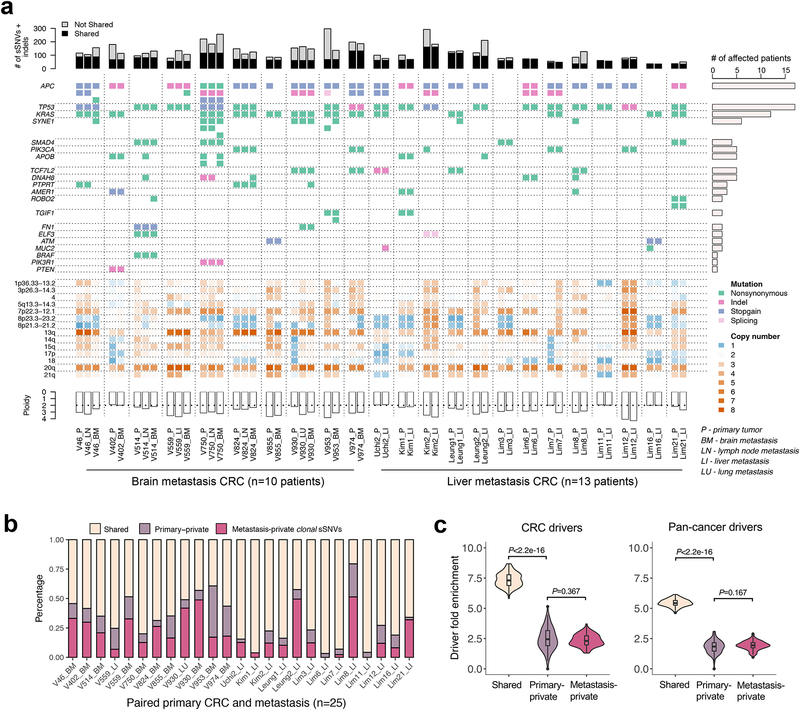
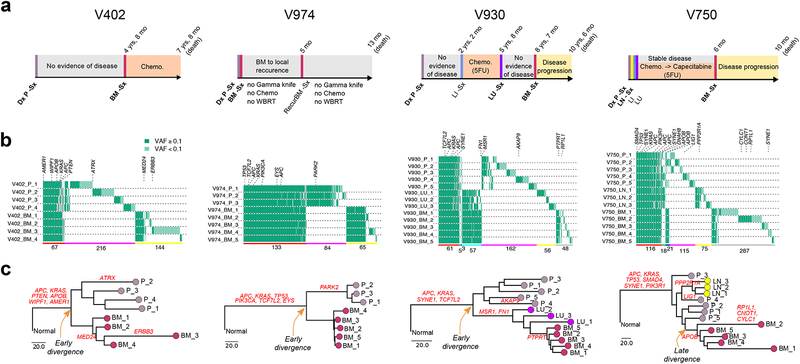
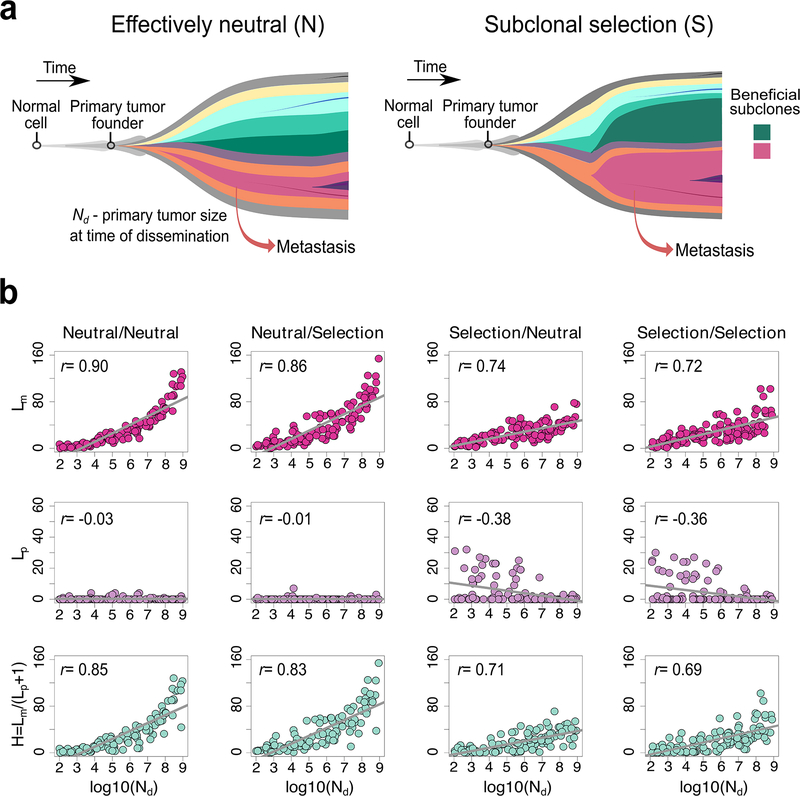
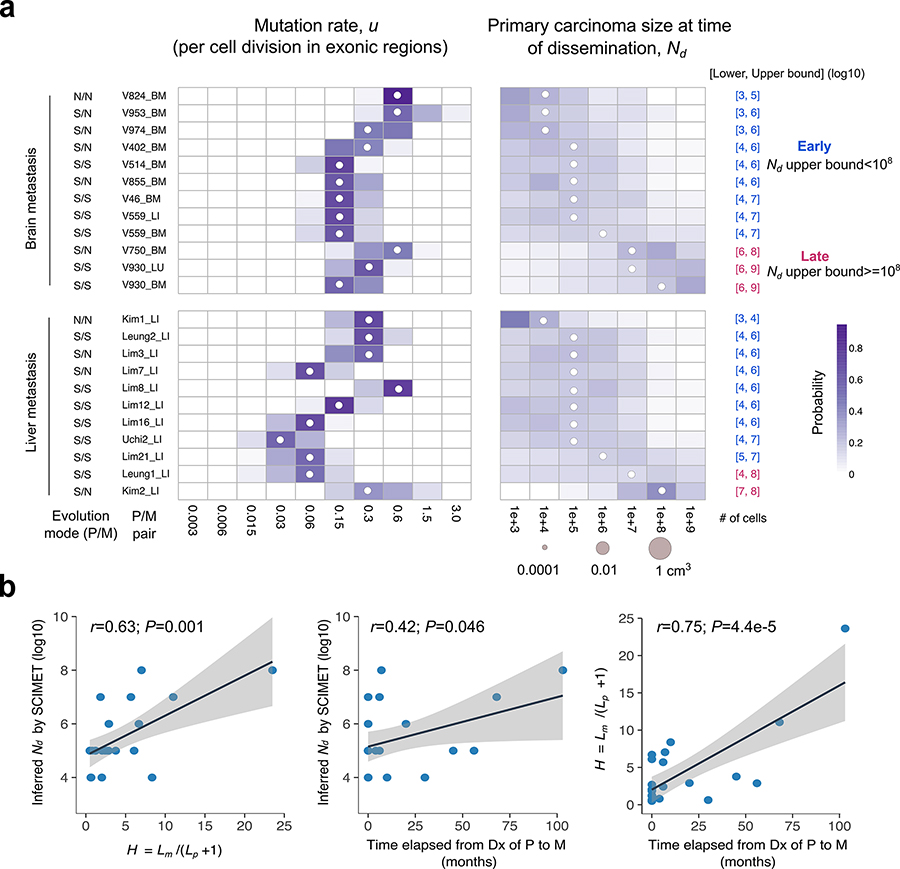
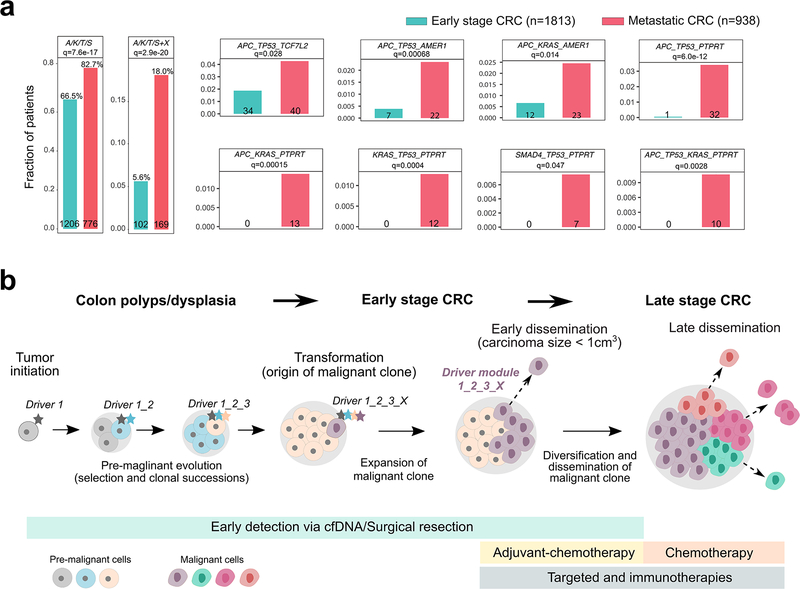
Both the timing and molecular determinants of metastasis are unknown, hindering treatment and prevention efforts. Here we characterize the evolutionary dynamics of this lethal process by analyzing exome-sequencing data from 118 biopsies from 23 patients with colorectal cancer with metastases to the liver or brain. The data show that the genomic divergence between the primary tumor and metastasis is low and that canonical driver genes were acquired early. Analysis within a spatial tumor growth model and statistical inference framework indicates that early disseminated cells commonly (81%, 17 out of 21 evaluable patients) seed metastases while the carcinoma is clinically undetectable (typically, less than 0.01 cm3). We validated the association between early drivers and metastasis in an independent cohort of 2,751 colorectal cancers, demonstrating their utility as biomarkers of metastasis. This conceptual and analytical framework provides quantitative in vivo evidence that systemic spread can occur early in colorectal cancer and illuminates strategies for patient stratification and therapeutic targeting of the canonical drivers of tumorigenesis.
Figures
Comment in
-
Does early metastatic seeding occur in colorectal cancer?Nat Rev Gastroenterol Hepatol. 2019 Nov;16(11):651-653. doi: 10.1038/s41575-019-0200-4. Nat Rev Gastroenterol Hepatol. 2019. PMID: 31417195 No abstract available.
References
-
- Turajlic S & Swanton C Metastasis as an evolutionary process. Science 352, 169–75 (2016). - PubMed
Methods-only References
-
- Berghoff AS et al. Differential role of angiogenesis and tumour cell proliferation in brain metastases according to primary tumour type: analysis of 639 cases. Neuropathol Appl Neurobiol 41, e41–55 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
