

Determining protein structures using deep mutagenesis
- PMID: 31209395
- PMCID: PMC7610650
- DOI: 10.1038/s41588-019-0431-x
Determining protein structures using deep mutagenesis
Abstract
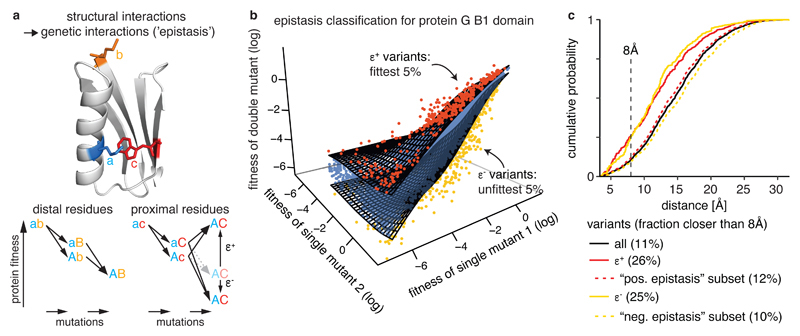
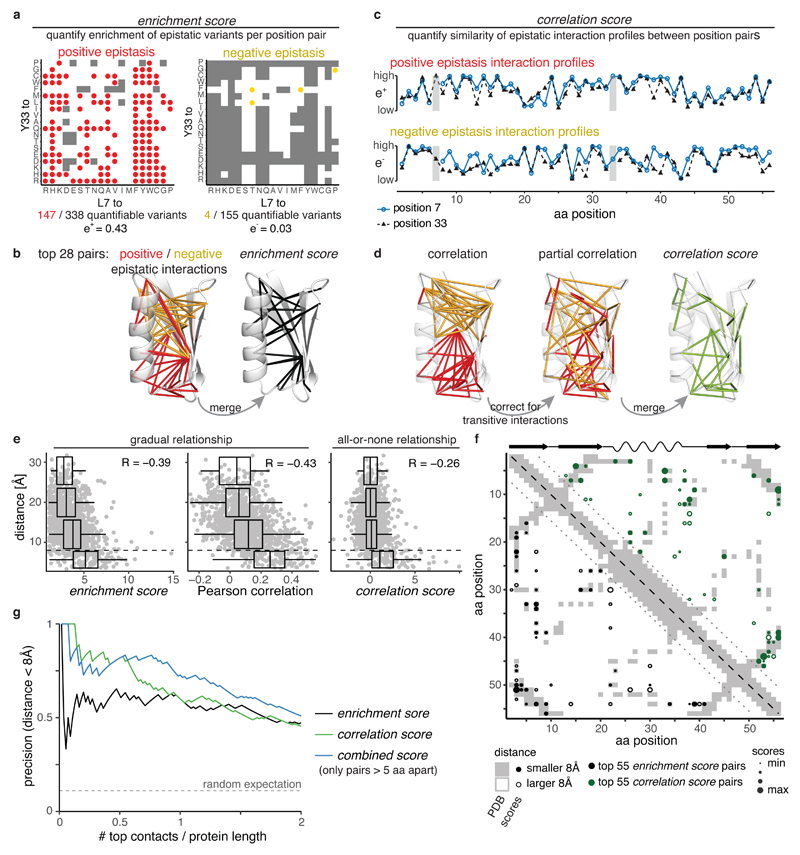
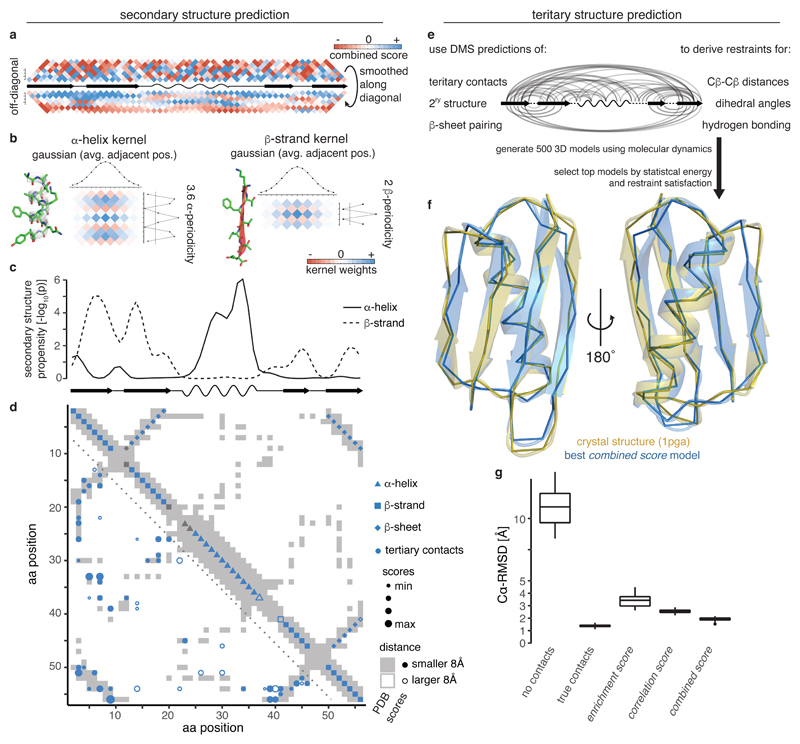
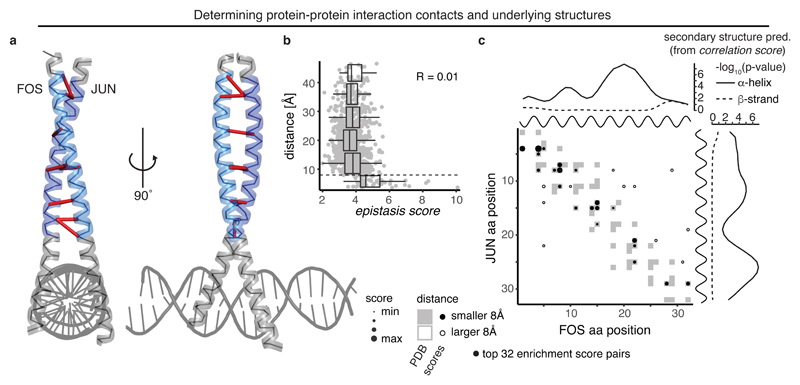
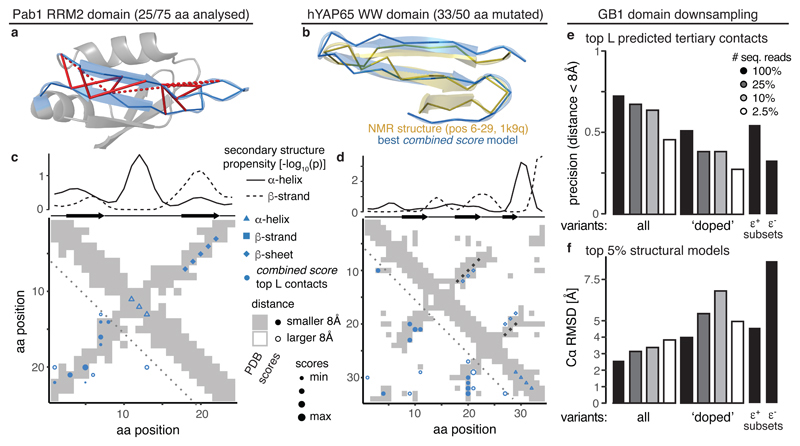
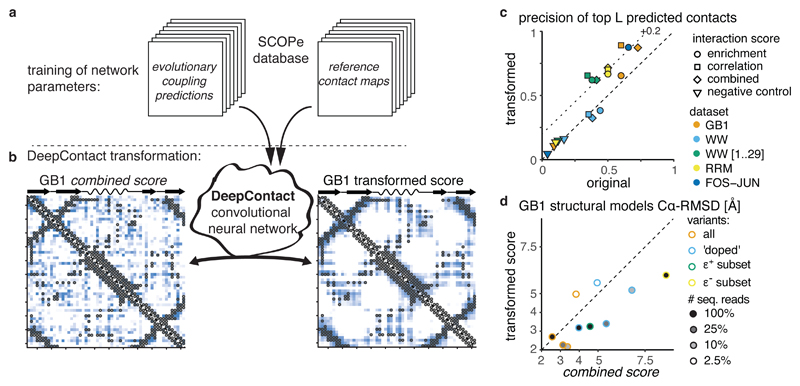
Determining the three-dimensional structures of macromolecules is a major goal of biological research, because of the close relationship between structure and function; however, thousands of protein domains still have unknown structures. Structure determination usually relies on physical techniques including X-ray crystallography, NMR spectroscopy and cryo-electron microscopy. Here we present a method that allows the high-resolution three-dimensional backbone structure of a biological macromolecule to be determined only from measurements of the activity of mutant variants of the molecule. This genetic approach to structure determination relies on the quantification of genetic interactions (epistasis) between mutations and the discrimination of direct from indirect interactions. This provides an alternative experimental strategy for structure determination, with the potential to reveal functional and in vivo structures.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
