

Toxin B Variants from Clostridium difficile Strains VPI 10463 and NAP1/027 Share Similar Substrate Profile and Cellular Intoxication Kinetics but Use Different Host Cell Entry Factors
- PMID: 31212980
- PMCID: PMC6628394
- DOI: 10.3390/toxins11060348
Toxin B Variants from Clostridium difficile Strains VPI 10463 and NAP1/027 Share Similar Substrate Profile and Cellular Intoxication Kinetics but Use Different Host Cell Entry Factors
Abstract
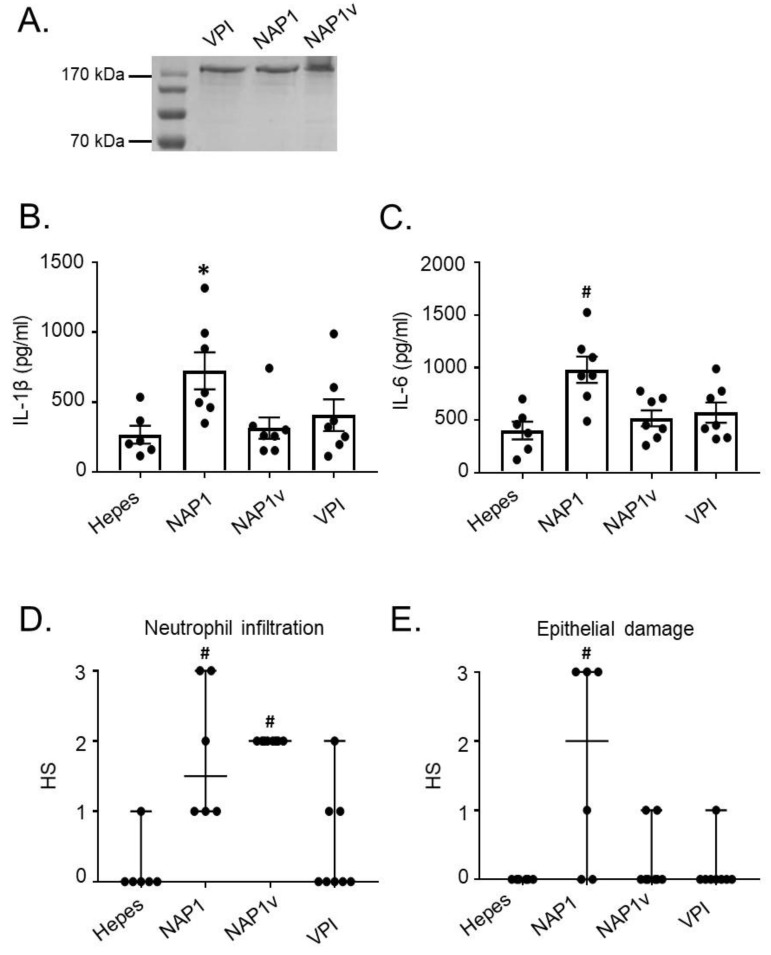
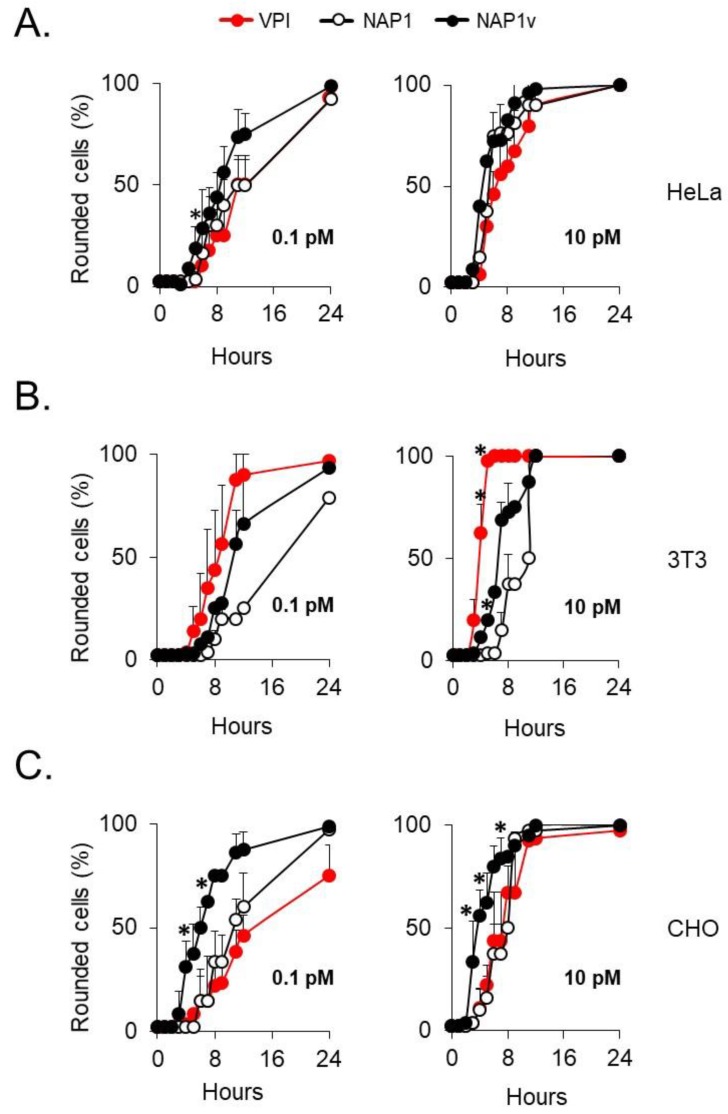
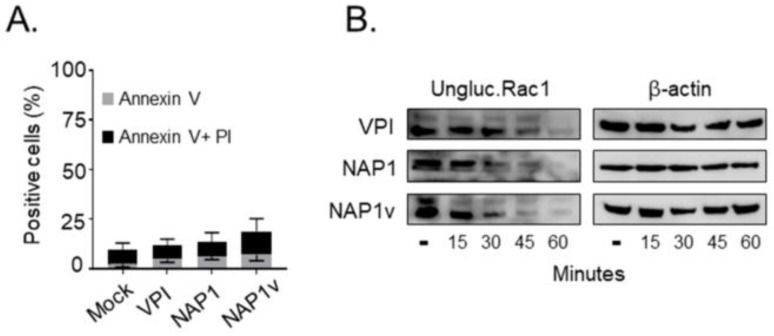
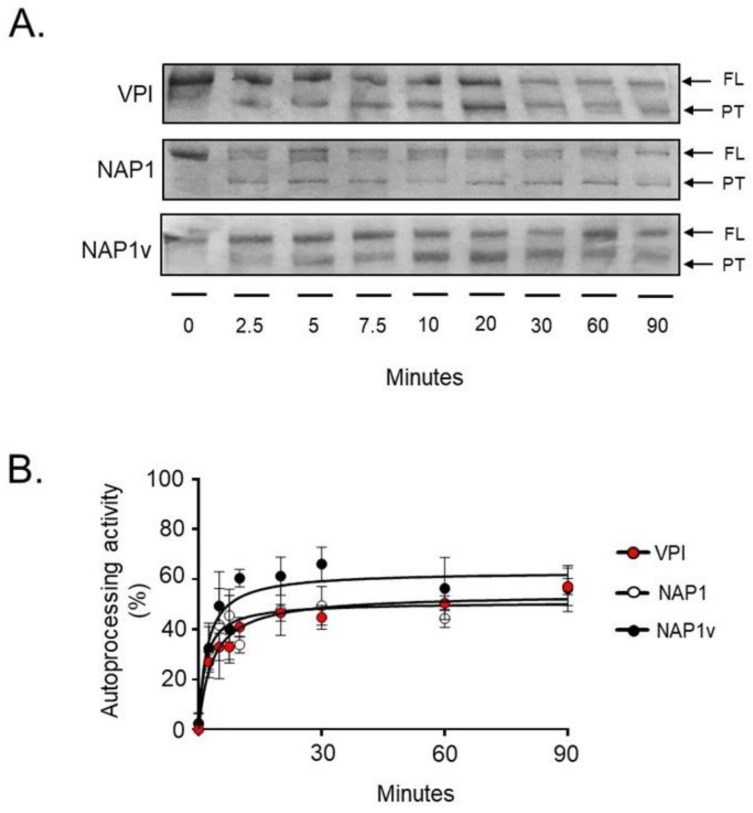
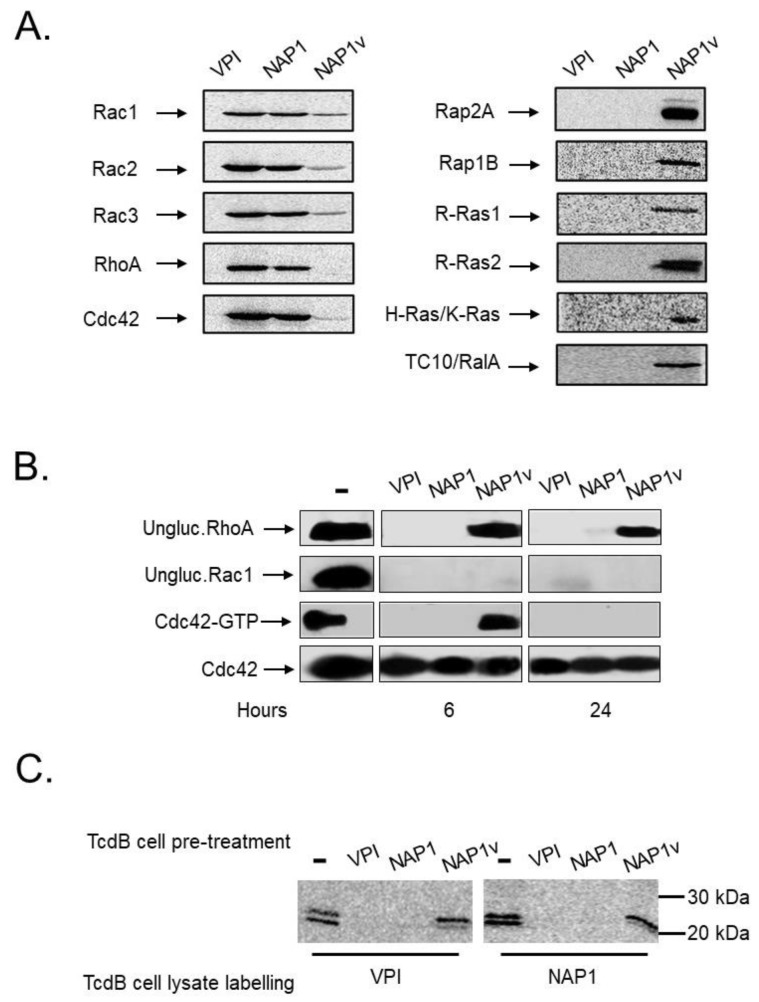
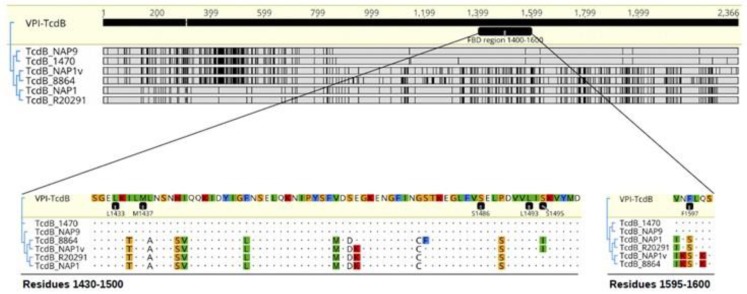
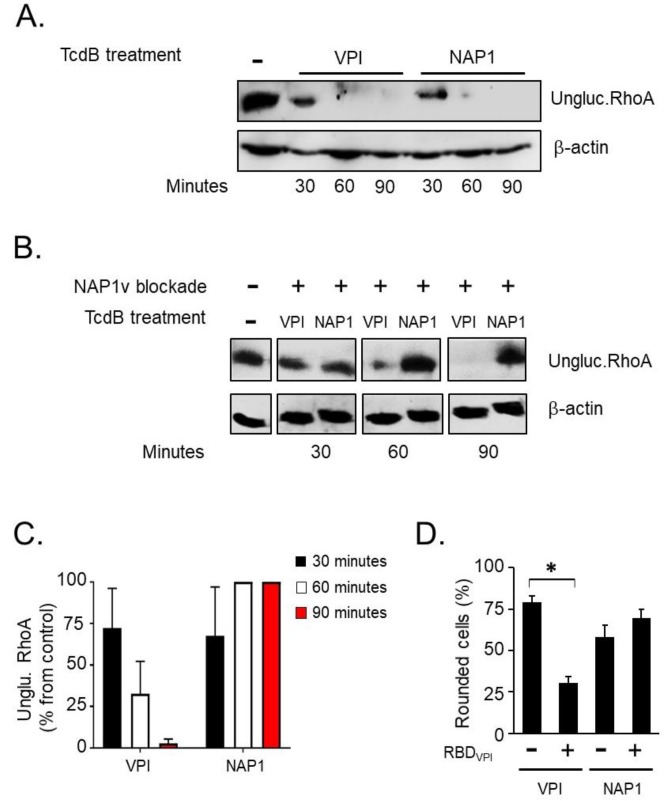
Clostridium difficile induces antibiotic-associated diarrhea due to the release of toxin A (TcdA) and toxin B (TcdB), the latter being its main virulence factor. The epidemic strain NAP1/027 has an increased virulence attributed to different factors. We compared cellular intoxication by TcdBNAP1 with that by the reference strain VPI 10463 (TcdBVPI). In a mouse ligated intestinal loop model, TcdBNAP1 induced higher neutrophil recruitment, cytokine release, and epithelial damage than TcdBVPI. Both toxins modified the same panel of small GTPases and exhibited similar in vitro autoprocessing kinetics. On the basis of sequence variations in the frizzled-binding domain (FBD), we reasoned that TcdBVPI and TcdBNAP1 might have different receptor specificities. To test this possibility, we used a TcdB from a NAP1 variant strain (TcdBNAP1v) unable to glucosylate RhoA but with the same receptor-binding domains as TcdBNAP1. Cells were preincubated with TcdBNAP1v to block cellular receptors, prior to intoxication with either TcdBVPI or TcdBNAP1. Preincubation with TcdBNAP1v blocked RhoA glucosylation by TcdBNAP1 but not by TcdBVPI, indicating that the toxins use different host factors for cell entry. This crucial difference might explain the increased biological activity of TcdBNAP1 in the intestine, representing a contributing factor for the increased virulence of the NAP1/027 strain.
Keywords: Clostridium difficile; NAP1/027 toxin B; frizzled receptors; receptor binding.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Analysis of TcdB Proteins within the Hypervirulent Clade 2 Reveals an Impact of RhoA Glucosylation on Clostridium difficile Proinflammatory Activities.Infect Immun. 2016 Jan 11;84(3):856-65. doi: 10.1128/IAI.01291-15. Infect Immun. 2016. PMID: 26755157 Free PMC article.
-
Defining the Roles of TcdA and TcdB in Localized Gastrointestinal Disease, Systemic Organ Damage, and the Host Response during Clostridium difficile Infections.mBio. 2015 Jun 2;6(3):e00551. doi: 10.1128/mBio.00551-15. mBio. 2015. PMID: 26037121 Free PMC article.
-
Clostridium difficile Toxin Biology.Annu Rev Microbiol. 2017 Sep 8;71:281-307. doi: 10.1146/annurev-micro-090816-093458. Epub 2017 Jun 28. Annu Rev Microbiol. 2017. PMID: 28657883 Review.
-
The Role of Rho GTPases in Toxicity of Clostridium difficile Toxins.Toxins (Basel). 2015 Dec 2;7(12):5254-67. doi: 10.3390/toxins7124874. Toxins (Basel). 2015. PMID: 26633511 Free PMC article. Review.
-
Clostridium difficile chimeric toxin receptor binding domain vaccine induced protection against different strains in active and passive challenge models.Vaccine. 2017 Jul 24;35(33):4079-4087. doi: 10.1016/j.vaccine.2017.06.062. Epub 2017 Jun 29. Vaccine. 2017. PMID: 28669616
Cited by
-
Clostridioides difficile infection damages colonic stem cells via TcdB, impairing epithelial repair and recovery from disease.Proc Natl Acad Sci U S A. 2020 Apr 7;117(14):8064-8073. doi: 10.1073/pnas.1915255117. Epub 2020 Mar 20. Proc Natl Acad Sci U S A. 2020. PMID: 32198200 Free PMC article.
-
Structural basis for CSPG4 as a receptor for TcdB and a therapeutic target in Clostridioides difficile infection.Nat Commun. 2021 Jun 18;12(1):3748. doi: 10.1038/s41467-021-23878-3. Nat Commun. 2021. PMID: 34145250 Free PMC article.
-
C. difficile intoxicates neurons and pericytes to drive neurogenic inflammation.Nature. 2023 Oct;622(7983):611-618. doi: 10.1038/s41586-023-06607-2. Epub 2023 Sep 12. Nature. 2023. PMID: 37699522 Free PMC article.
-
Designed Ankyrin Repeat Protein (DARPin) Neutralizers of TcdB from Clostridium difficile Ribotype 027.mSphere. 2019 Oct 2;4(5):e00596-19. doi: 10.1128/mSphere.00596-19. mSphere. 2019. PMID: 31578248 Free PMC article.
-
Exploring the Toxin-Mediated Mechanisms in Clostridioides difficile Infection.Microorganisms. 2024 May 16;12(5):1004. doi: 10.3390/microorganisms12051004. Microorganisms. 2024. PMID: 38792835 Free PMC article. Review.
References
-
- Carter G.P., Chakravorty A., Pham Nguyen T.A., Mileto S., Schreiber F., Li L., Howarth P., Clare S., Cunningham B., Sambol S.P., et al. Defining the roles of TcdA and TcdB in localized gastrointestinal disease, systemic organ damage, and the host response during Clostridium difficile Infections. mBio. 2015;6:e00551-15. doi: 10.1128/mBio.00551-15. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
