

Inherited IL-18BP deficiency in human fulminant viral hepatitis
- PMID: 31213488
- PMCID: PMC6683989
- DOI: 10.1084/jem.20190669
Inherited IL-18BP deficiency in human fulminant viral hepatitis
Abstract
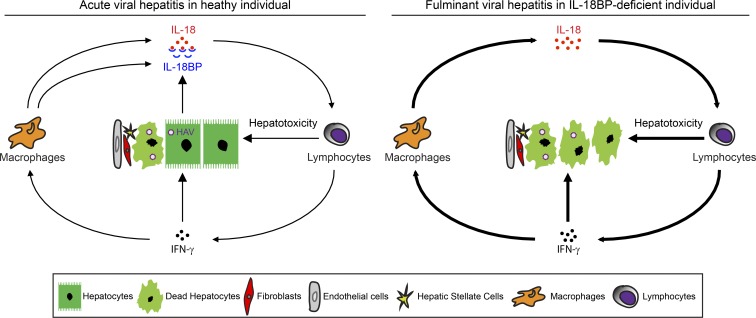
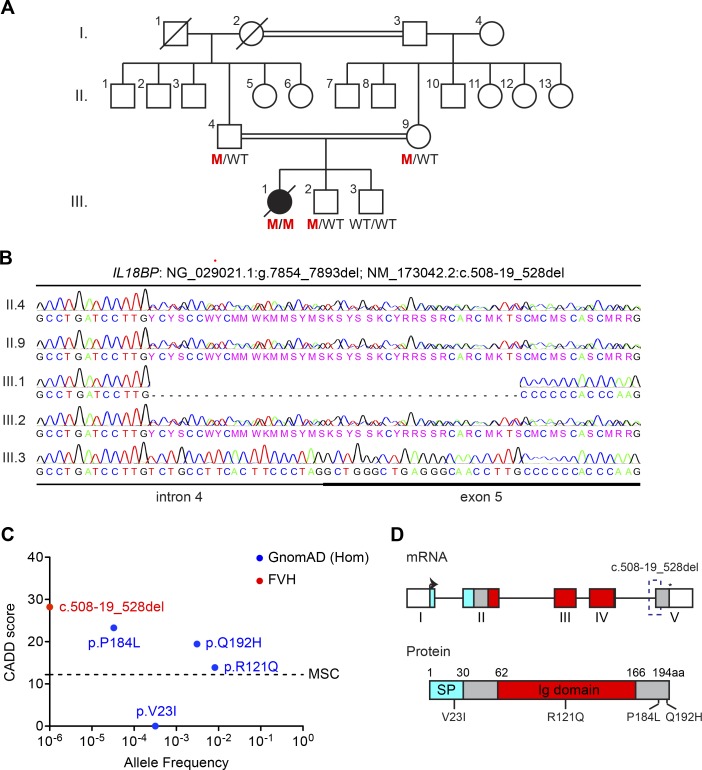
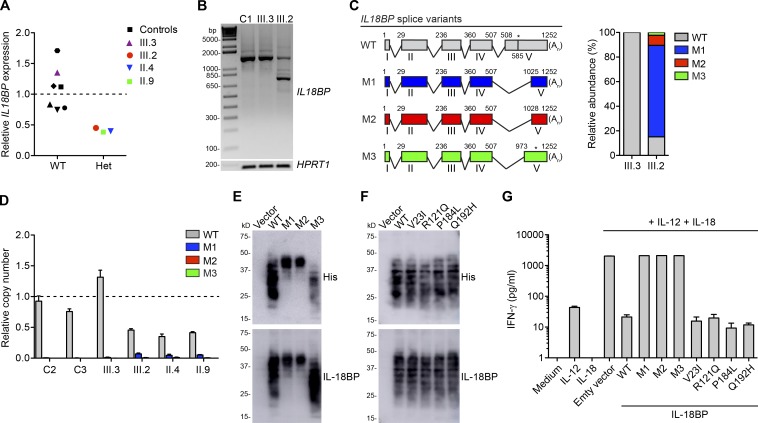
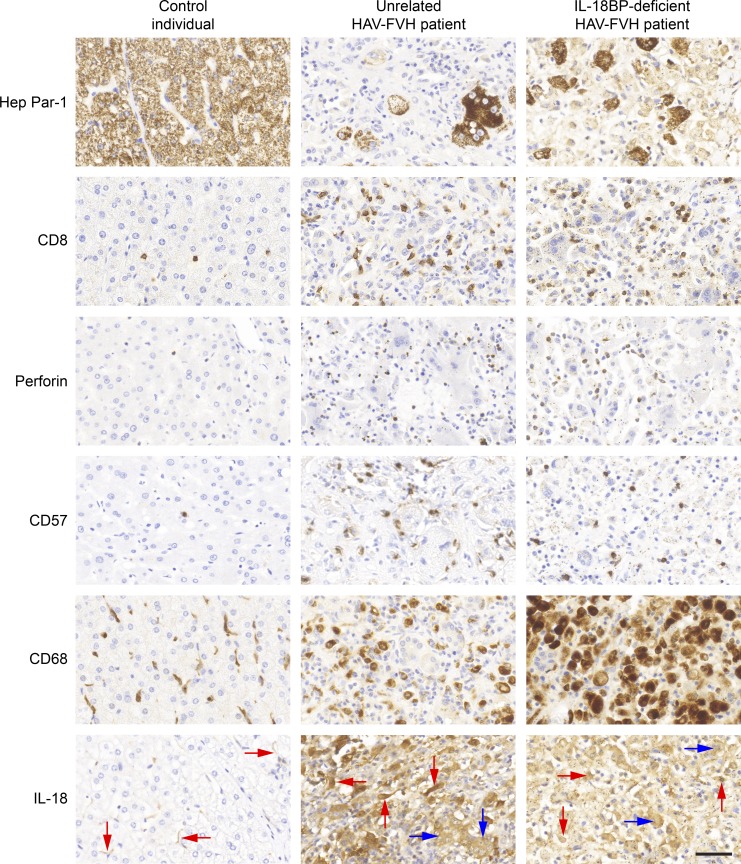
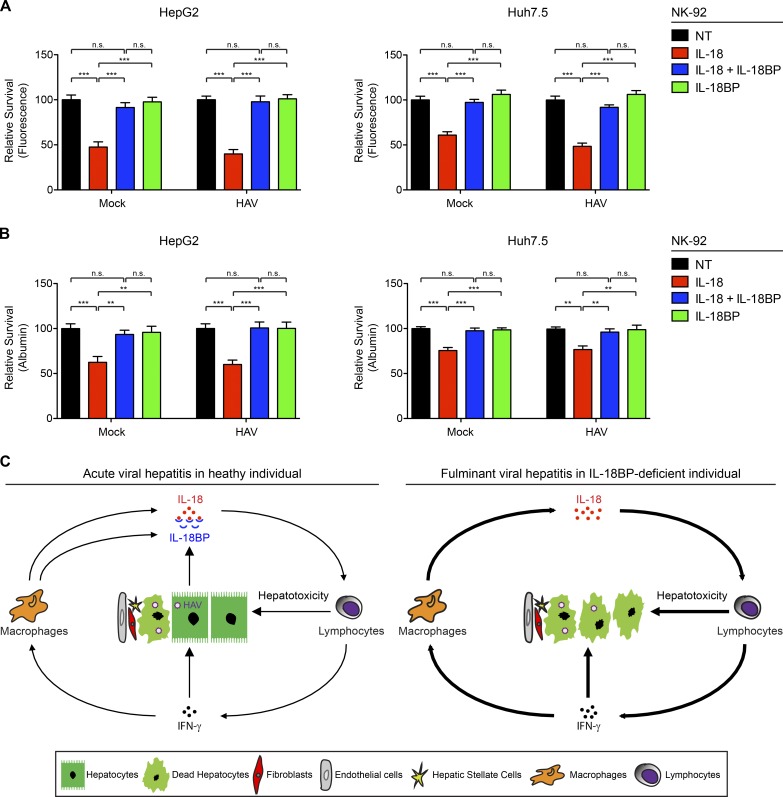
Fulminant viral hepatitis (FVH) is a devastating and unexplained condition that strikes otherwise healthy individuals during primary infection with common liver-tropic viruses. We report a child who died of FVH upon infection with hepatitis A virus (HAV) at age 11 yr and who was homozygous for a private 40-nucleotide deletion in IL18BP, which encodes the IL-18 binding protein (IL-18BP). This mutation is loss-of-function, unlike the variants found in a homozygous state in public databases. We show that human IL-18 and IL-18BP are both secreted mostly by hepatocytes and macrophages in the liver. Moreover, in the absence of IL-18BP, excessive NK cell activation by IL-18 results in uncontrolled killing of human hepatocytes in vitro. Inherited human IL-18BP deficiency thus underlies fulminant HAV hepatitis by unleashing IL-18. These findings provide proof-of-principle that FVH can be caused by single-gene inborn errors that selectively disrupt liver-specific immunity. They also show that human IL-18 is toxic to the liver and that IL-18BP is its antidote.
© 2019 Belkaya et al.
Figures
Comment in
-
No IL-18BP? Avoid HAV.J Exp Med. 2019 Aug 5;216(8):1728-1729. doi: 10.1084/jem.20190841. Epub 2019 Jun 26. J Exp Med. 2019. PMID: 31243052 Free PMC article.
References
-
- Ajmera V., Xia G., Vaughan G., Forbi J.C., Ganova-Raeva L.M., Khudyakov Y., Opio C.K., Taylor R., Restrepo R., Munoz S., et al. Acute Liver Failure Study Group . 2011. What factors determine the severity of hepatitis A-related acute liver failure? J. Viral Hepat. 18:e167–e174. 10.1111/j.1365-2893.2010.01410.x - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
