

SHOC2 complex-driven RAF dimerization selectively contributes to ERK pathway dynamics
- PMID: 31213532
- PMCID: PMC6613145
- DOI: 10.1073/pnas.1902658116
SHOC2 complex-driven RAF dimerization selectively contributes to ERK pathway dynamics
Abstract
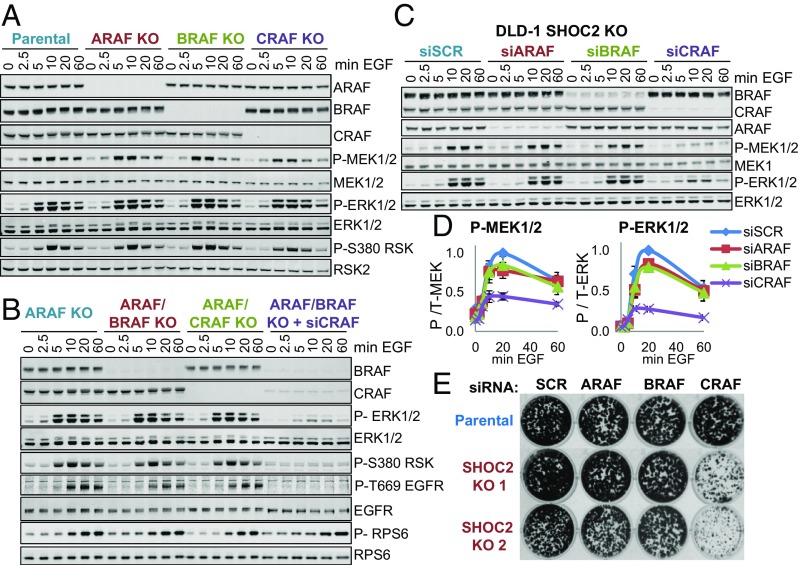
Despite the crucial role of RAF kinases in cell signaling and disease, we still lack a complete understanding of their regulation. Heterodimerization of RAF kinases as well as dephosphorylation of a conserved "S259" inhibitory site are important steps for RAF activation but the precise mechanisms and dynamics remain unclear. A ternary complex comprised of SHOC2, MRAS, and PP1 (SHOC2 complex) functions as a RAF S259 holophosphatase and gain-of-function mutations in SHOC2, MRAS, and PP1 that promote complex formation are found in Noonan syndrome. Here we show that SHOC2 complex-mediated S259 RAF dephosphorylation is critically required for growth factor-induced RAF heterodimerization as well as for MEK dissociation from BRAF. We also uncover SHOC2-independent mechanisms of RAF and ERK pathway activation that rely on N-region phosphorylation of CRAF. In DLD-1 cells stimulated with EGF, SHOC2 function is essential for a rapid transient phase of ERK activation, but is not required for a slow, sustained phase that is instead driven by palmitoylated H/N-RAS proteins and CRAF. Whereas redundant SHOC2-dependent and -independent mechanisms of RAF and ERK activation make SHOC2 dispensable for proliferation in 2D, KRAS mutant cells preferentially rely on SHOC2 for ERK signaling under anchorage-independent conditions. Our study highlights a context-dependent contribution of SHOC2 to ERK pathway dynamics that is preferentially engaged by KRAS oncogenic signaling and provides a biochemical framework for selective ERK pathway inhibition by targeting the SHOC2 holophosphatase.
Keywords: ERK; MRAS; RAF; RAS; SHOC2.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
