

Lysosomal acid lipase deficiency - early diagnosis is the key
- PMID: 31213932
- PMCID: PMC6536894
- DOI: 10.2147/HMER.S201630
Lysosomal acid lipase deficiency - early diagnosis is the key
Abstract
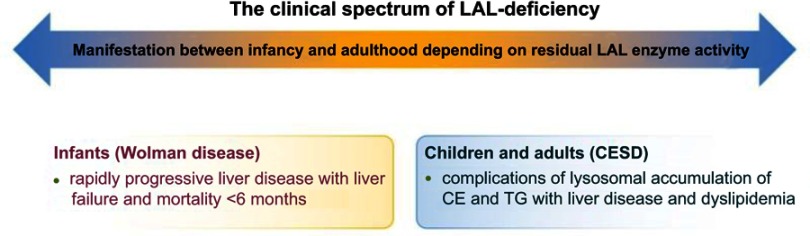
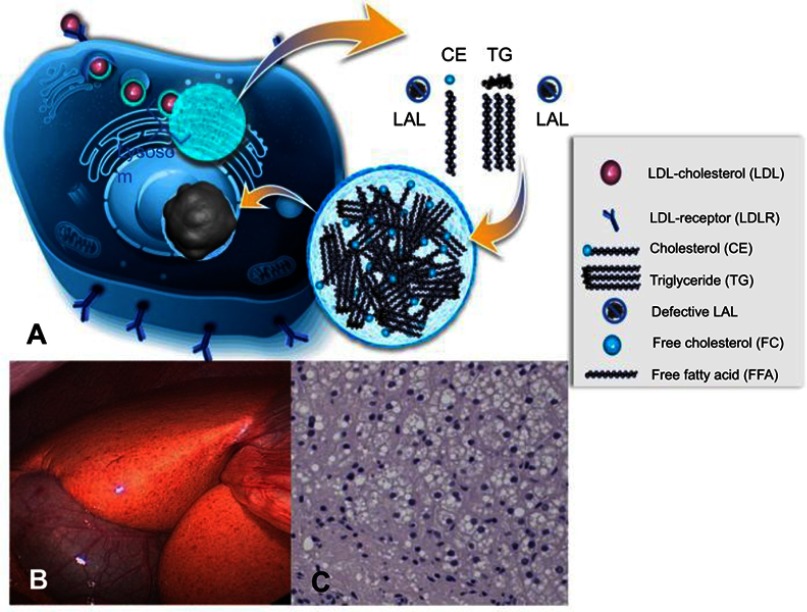
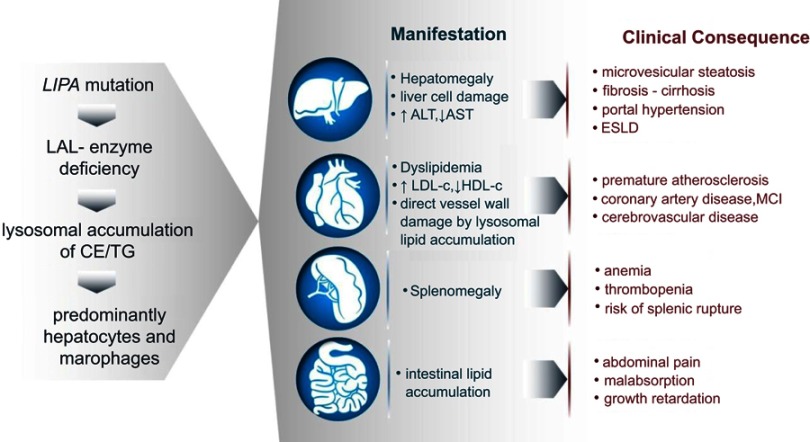
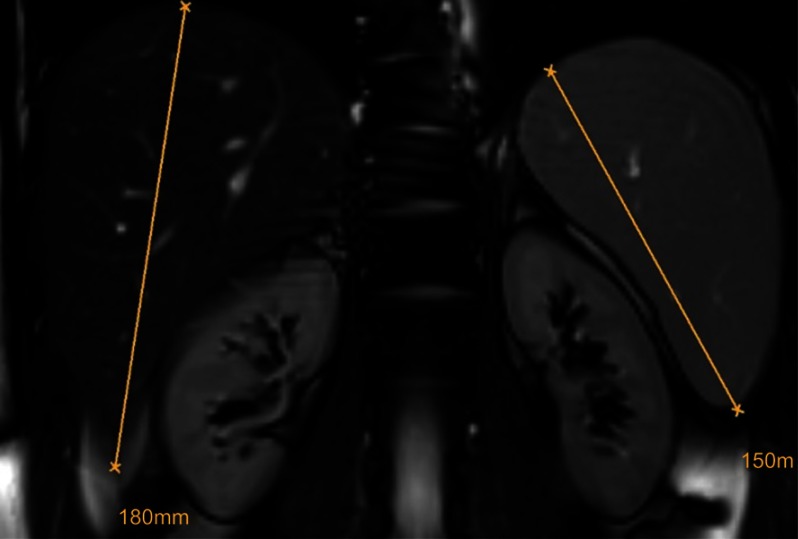
Lysosomal acid lipase deficiency (LAL-D) is an ultra-rare lysosomal storage disease that may present from infancy to late adulthood depending on residual enzyme activity. While the severe form manifests as a rapidly progressive disease with near universal mortality within the first 6 months of life, milder forms frequently go undiagnosed for prolonged periods and typically present with progressive fatty liver disease, enlarged spleen, atherogenic dyslipidemia and premature atherosclerosis. The adult variant of LAL-D is typically diagnosed late or even overlooked due to the unspecific nature of the presenting symptoms, which are similar to common changes observed in the context of the metabolic syndrome. This review is aimed at delineating clinically useful scenarios in which pediatric or adult medicine clinicians should be aware of LAL-D as a differential diagnosis for selected patients. This is particularly relevant as a potentially life-saving enzyme replacement therapy has become available and the diagnosis can easily be ruled out or confirmed using a dried blood spot test.
Keywords: atherogenic dyslipidemia; liver cirrhosis; low HDL; lysosomal acid lipase; microvesicular steatosis.
Conflict of interest statement
Elmar Aigner, MD, has received consulting and speaking honoraria from Alexion Pharmaceuticals. The other authors report no conflicts of interest in this work.
Figures
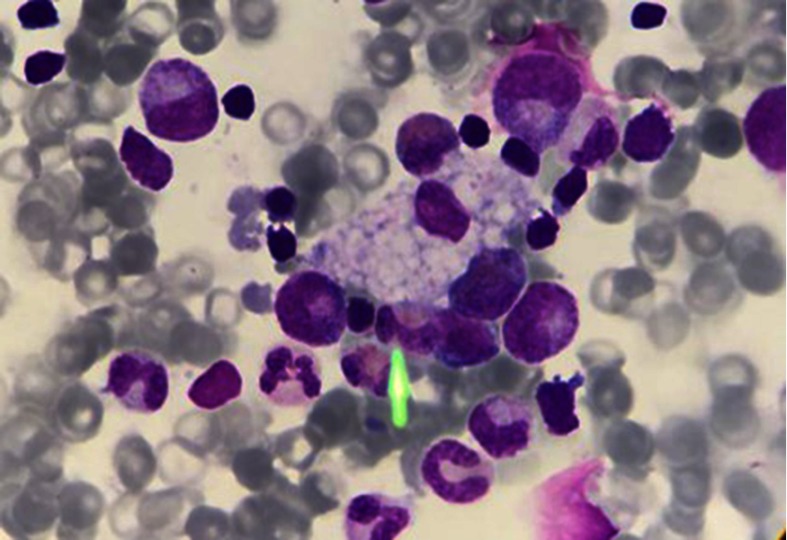
References
-
- Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J Biol Chem. 1975;250(21):8487–8495. - PubMed
-
- Beaudet AL, Ferry GD, Nichols BL Jr, Rosenberg HS. Cholesterol ester storage disease: clinical, biochemical, and pathological studies. J Pediatr. 1977;90(6):910–914. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
