

MSH3 modifies somatic instability and disease severity in Huntington's and myotonic dystrophy type 1
- PMID: 31216018
- PMCID: PMC6598626
- DOI: 10.1093/brain/awz115
MSH3 modifies somatic instability and disease severity in Huntington's and myotonic dystrophy type 1
Abstract
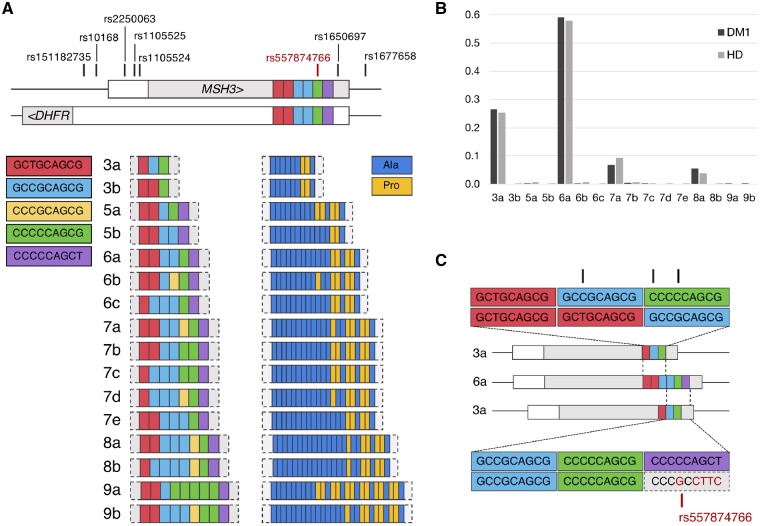
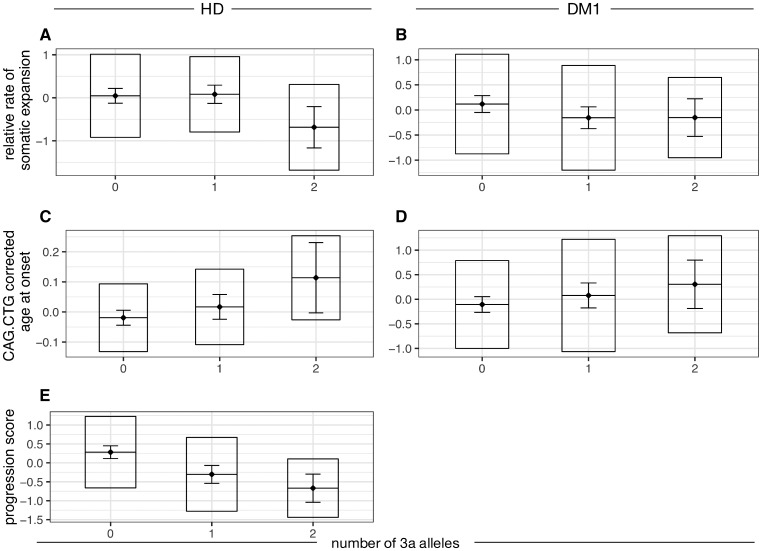
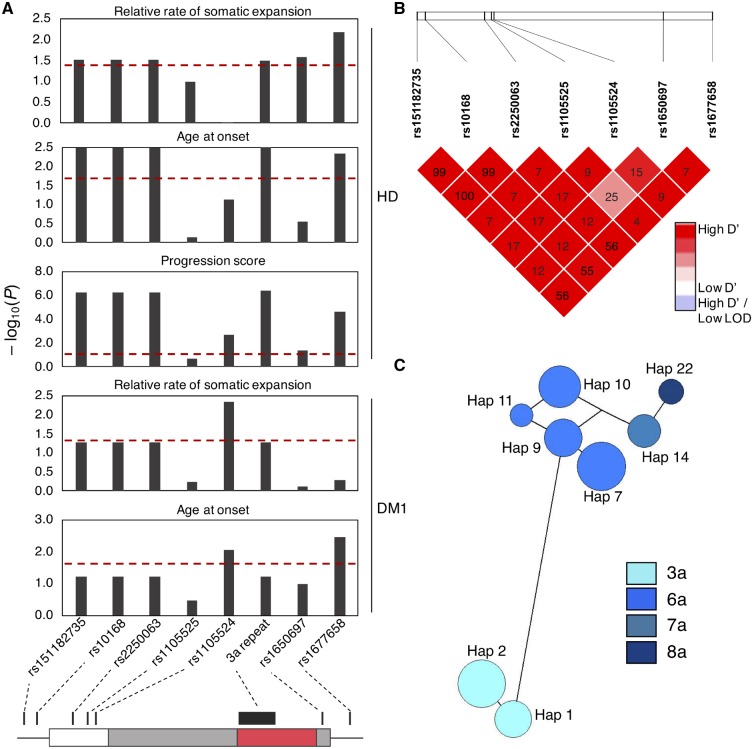
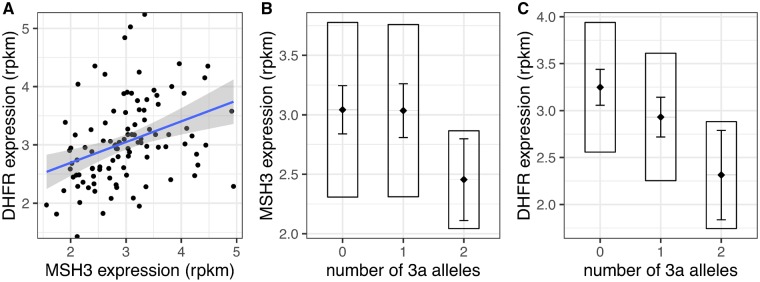
The mismatch repair gene MSH3 has been implicated as a genetic modifier of the CAG·CTG repeat expansion disorders Huntington's disease and myotonic dystrophy type 1. A recent Huntington's disease genome-wide association study found rs557874766, an imputed single nucleotide polymorphism located within a polymorphic 9 bp tandem repeat in MSH3/DHFR, as the variant most significantly associated with progression in Huntington's disease. Using Illumina sequencing in Huntington's disease and myotonic dystrophy type 1 subjects, we show that rs557874766 is an alignment artefact, the minor allele for which corresponds to a three-repeat allele in MSH3 exon 1 that is associated with a reduced rate of somatic CAG·CTG expansion (P = 0.004) and delayed disease onset (P = 0.003) in both Huntington's disease and myotonic dystrophy type 1, and slower progression (P = 3.86 × 10-7) in Huntington's disease. RNA-Seq of whole blood in the Huntington's disease subjects found that repeat variants are associated with MSH3 and DHFR expression. A transcriptome-wide association study in the Huntington's disease cohort found increased MSH3 and DHFR expression are associated with disease progression. These results suggest that variation in the MSH3 exon 1 repeat region influences somatic expansion and disease phenotype in Huntington's disease and myotonic dystrophy type 1, and suggests a common DNA repair mechanism operates in both repeat expansion diseases.
Keywords: Huntington’s disease; association study; movement disorders; myotonic dystrophy; transcriptomics.
© The Author(s) (2019). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
Comment in
-
The repeat variant in MSH3 is not a genetic modifier for spinocerebellar ataxia type 3 and Friedreich's ataxia.Brain. 2020 Apr 1;143(4):e25. doi: 10.1093/brain/awaa043. Brain. 2020. PMID: 32154839 Free PMC article. No abstract available.
-
Reply: The repeat variant in MSH3 is not a genetic modifier for spinocerebellar ataxia type 3 and Friedreich's ataxia.Brain. 2020 Apr 1;143(4):e26. doi: 10.1093/brain/awaa044. Brain. 2020. PMID: 32154840 No abstract available.
References
-
- Ashizawa T, Dubel JR, Harati Y. Somatic instability of CTG repeat in myotonic dystrophy. Neurology 1993; 43: 2674–8. - PubMed
-
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 1999; 16: 37–48. - PubMed
-
- Bates G, Tabrizi S, Jones L. Huntington’s disease. Oxford: Oxford University Press; 2014.
-
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al.Huntington disease. Nat Rev Dis Primers 2015; 1: 15005. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
