

Two-Step Activation Mechanism of the ClpB Disaggregase for Sequential Substrate Threading by the Main ATPase Motor
- PMID: 31216466
- PMCID: PMC6593972
- DOI: 10.1016/j.celrep.2019.05.075
Two-Step Activation Mechanism of the ClpB Disaggregase for Sequential Substrate Threading by the Main ATPase Motor
Abstract
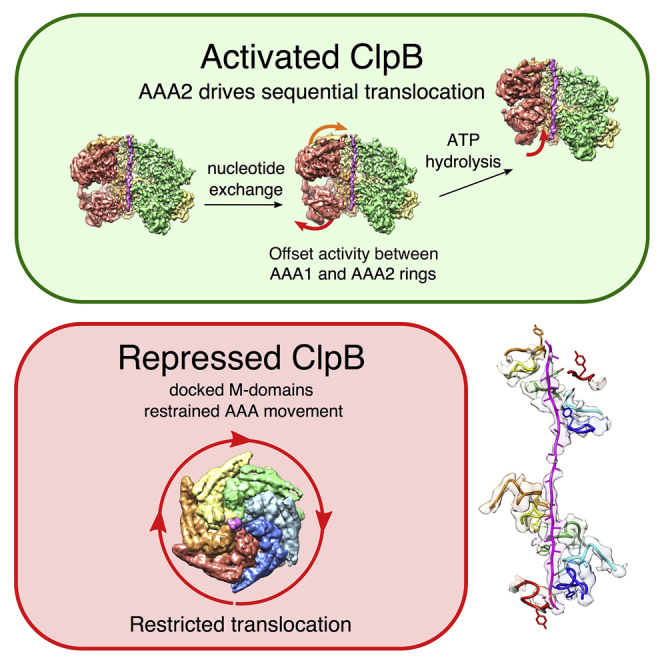
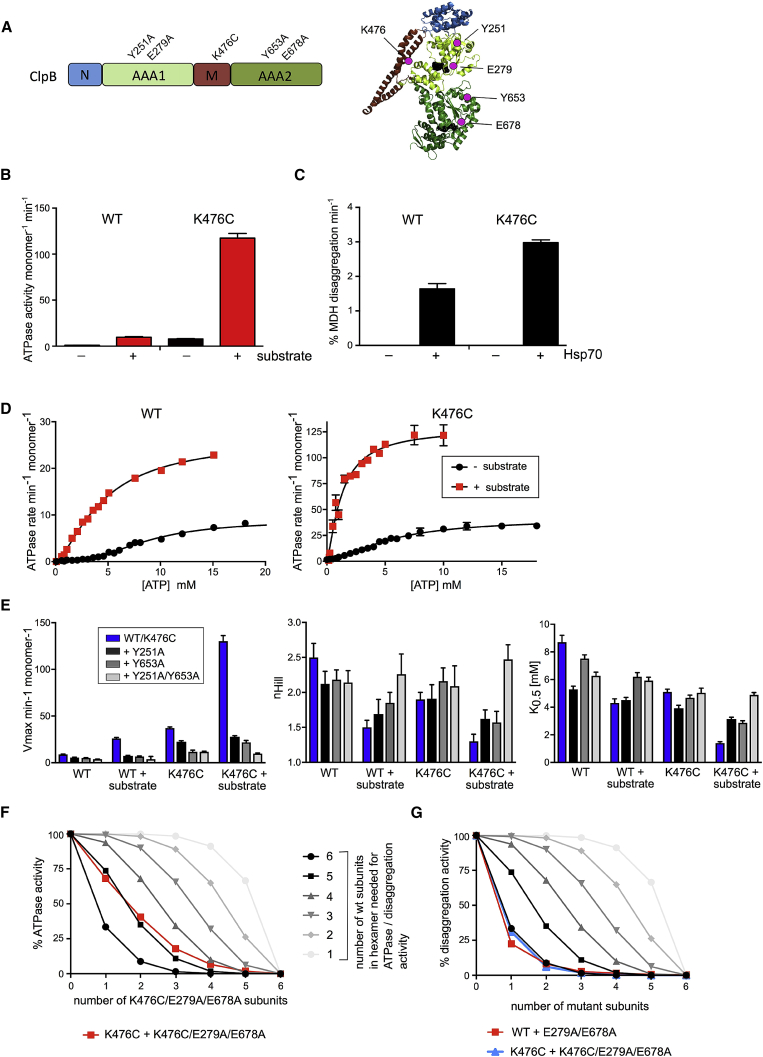
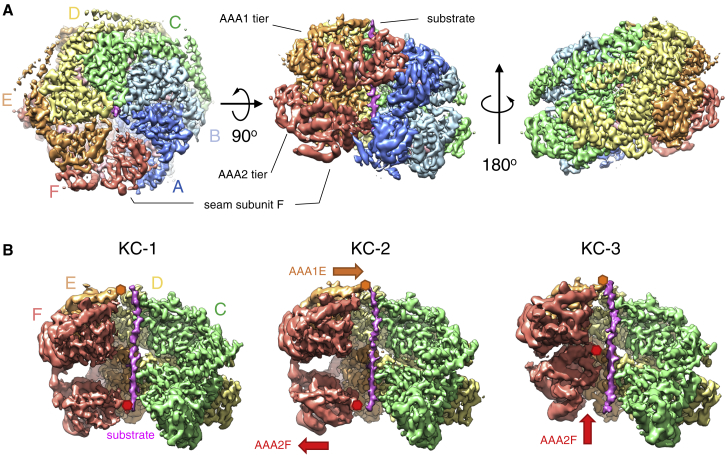
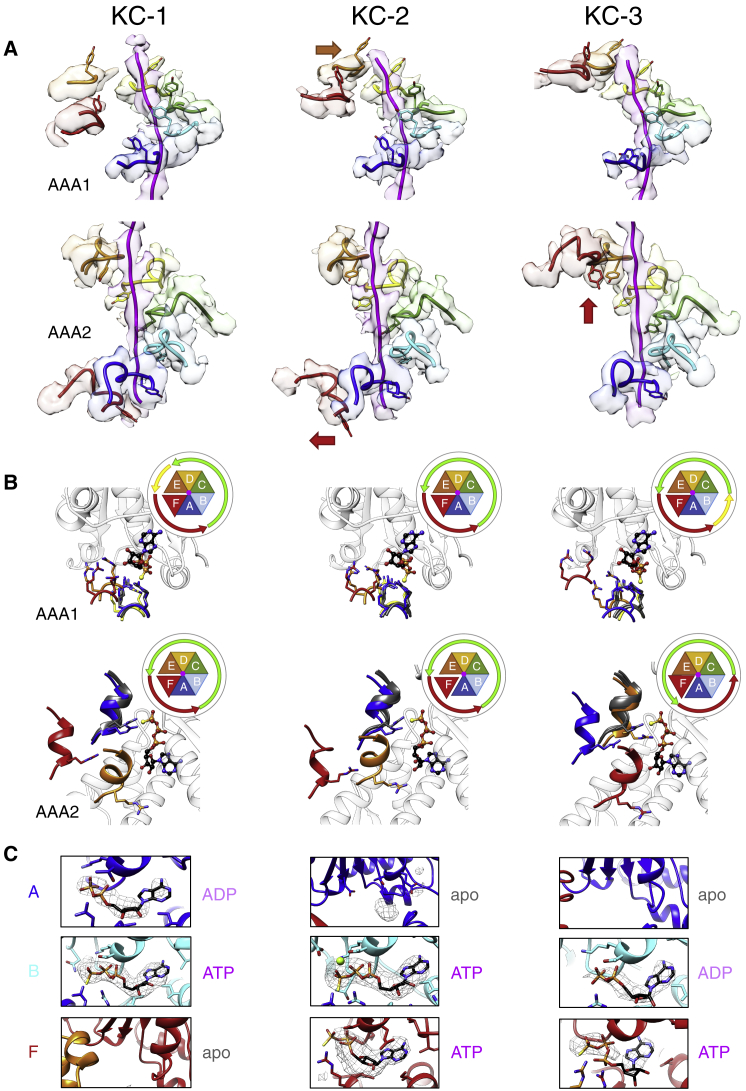
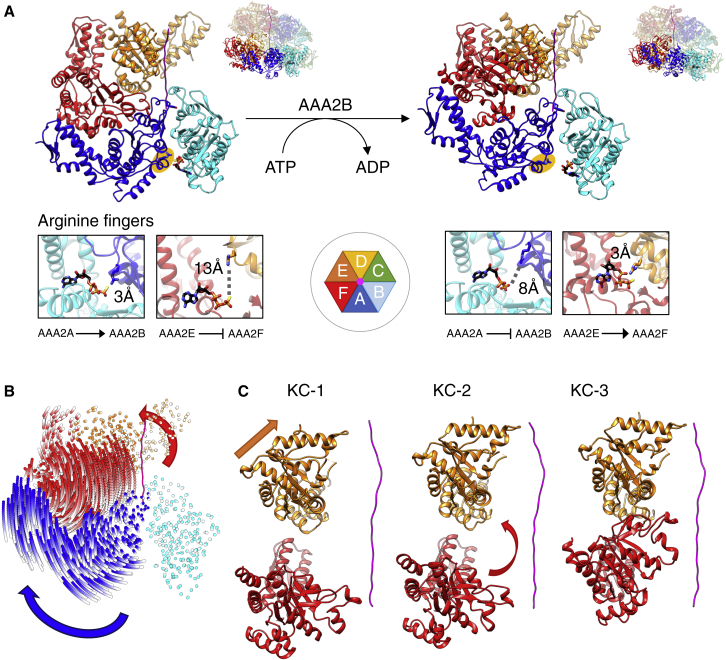
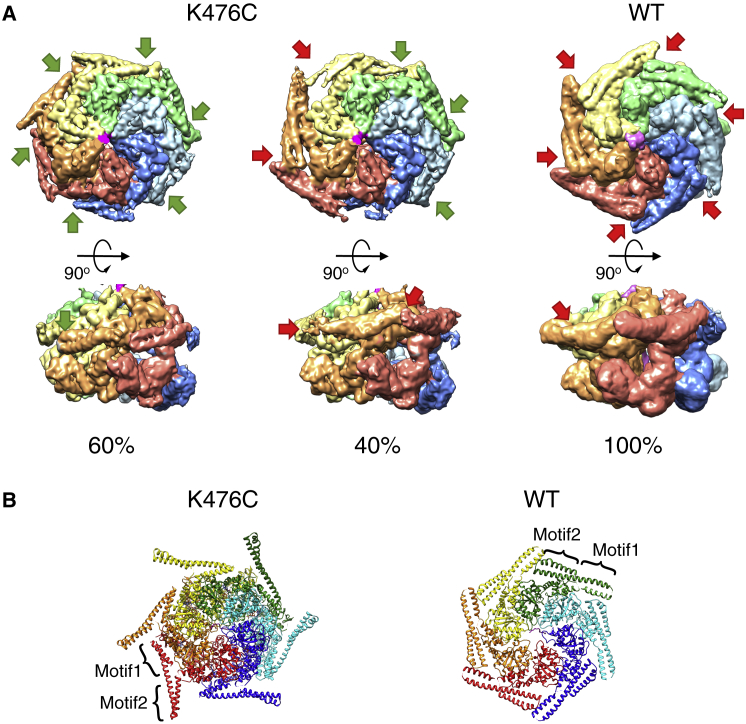
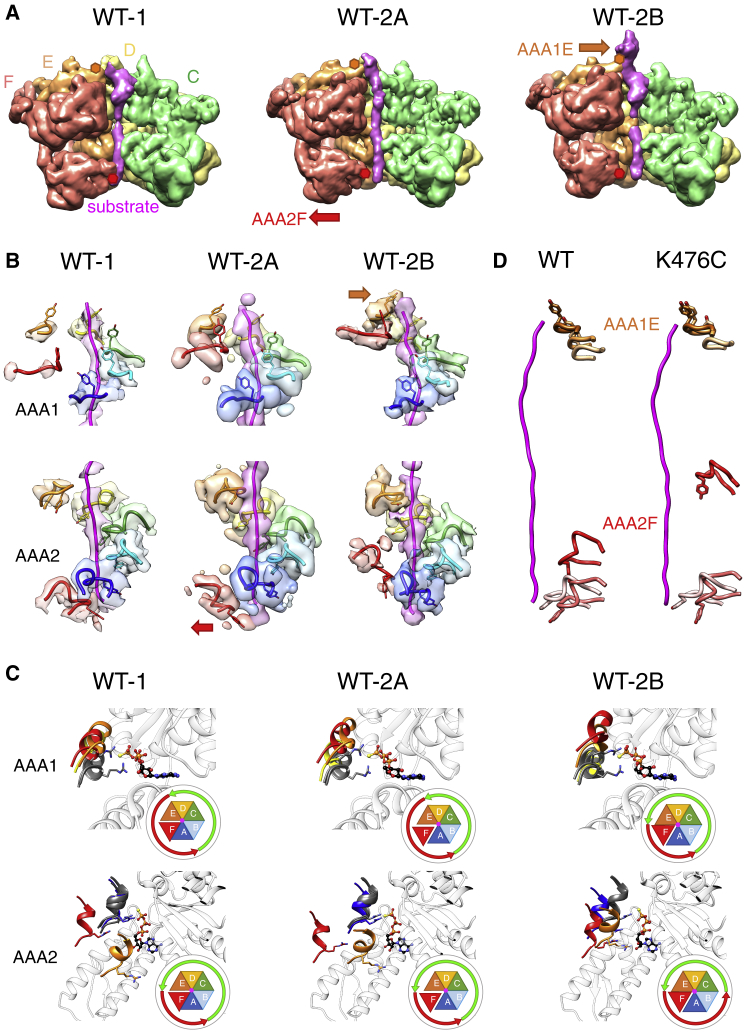
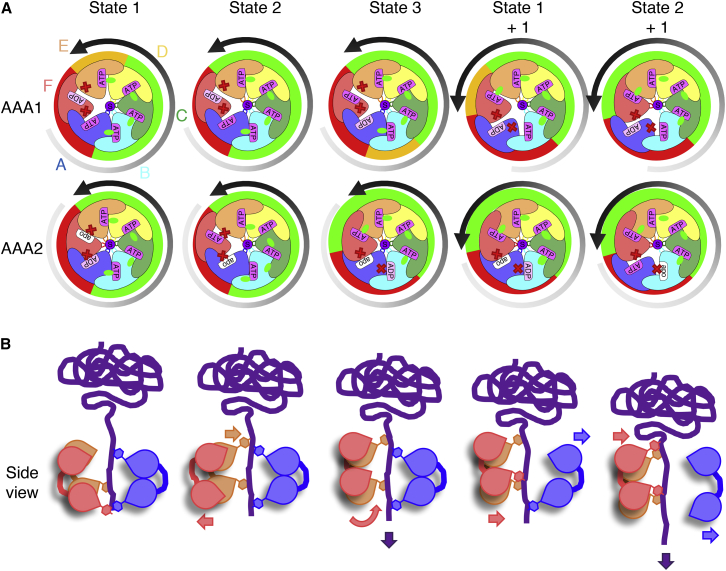
AAA+ proteins form asymmetric hexameric rings that hydrolyze ATP and thread substrate proteins through a central channel via mobile substrate-binding pore loops. Understanding how ATPase and threading activities are regulated and intertwined is key to understanding the AAA+ protein mechanism. We studied the disaggregase ClpB, which contains tandem ATPase domains (AAA1, AAA2) and shifts between low and high ATPase and threading activities. Coiled-coil M-domains repress ClpB activity by encircling the AAA1 ring. Here, we determine the mechanism of ClpB activation by comparing ATPase mechanisms and cryo-EM structures of ClpB wild-type and a constitutively active ClpB M-domain mutant. We show that ClpB activation reduces ATPase cooperativity and induces a sequential mode of ATP hydrolysis in the AAA2 ring, the main ATPase motor. AAA1 and AAA2 rings do not work synchronously but in alternating cycles. This ensures high grip, enabling substrate threading via a processive, rope-climbing mechanism.
Keywords: AAA+; Hsp100; chaperone; cryo-EM; protein disaggregation; protein unfolding.
Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Basic mechanism of the autonomous ClpG disaggregase.J Biol Chem. 2021 Jan-Jun;296:100460. doi: 10.1016/j.jbc.2021.100460. Epub 2021 Feb 24. J Biol Chem. 2021. PMID: 33639171 Free PMC article.
-
Structural basis for substrate gripping and translocation by the ClpB AAA+ disaggregase.Nat Commun. 2019 Jun 3;10(1):2393. doi: 10.1038/s41467-019-10150-y. Nat Commun. 2019. PMID: 31160557 Free PMC article.
-
Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity.J Biol Chem. 2003 May 16;278(20):17615-24. doi: 10.1074/jbc.M209686200. Epub 2003 Mar 6. J Biol Chem. 2003. PMID: 12624113
-
Molecular chaperones: structure of a protein disaggregase.Curr Biol. 2004 Jan 20;14(2):R78-80. doi: 10.1016/j.cub.2003.12.051. Curr Biol. 2004. PMID: 14738756 Review.
-
The elusive middle domain of Hsp104 and ClpB: location and function.Biochim Biophys Acta. 2012 Jan;1823(1):29-39. doi: 10.1016/j.bbamcr.2011.07.014. Epub 2011 Jul 24. Biochim Biophys Acta. 2012. PMID: 21843558 Free PMC article. Review.
Cited by
-
A Potential Mechanism for Targeting Aggregates With Proteasomes and Disaggregases in Liquid Droplets.Front Aging Neurosci. 2022 Apr 6;14:854380. doi: 10.3389/fnagi.2022.854380. eCollection 2022. Front Aging Neurosci. 2022. PMID: 35517053 Free PMC article. Review.
-
Basic mechanism of the autonomous ClpG disaggregase.J Biol Chem. 2021 Jan-Jun;296:100460. doi: 10.1016/j.jbc.2021.100460. Epub 2021 Feb 24. J Biol Chem. 2021. PMID: 33639171 Free PMC article.
-
Protein quality control: from mechanism to disease : EMBO Workshop, Costa de la Calma (Mallorca), Spain, April 28 - May 03, 2019.Cell Stress Chaperones. 2019 Nov;24(6):1013-1026. doi: 10.1007/s12192-019-01040-9. Epub 2019 Nov 11. Cell Stress Chaperones. 2019. PMID: 31713048 Free PMC article.
-
Stairway to translocation: AAA+ motor structures reveal the mechanisms of ATP-dependent substrate translocation.Protein Sci. 2020 Feb;29(2):407-419. doi: 10.1002/pro.3743. Epub 2019 Oct 17. Protein Sci. 2020. PMID: 31599052 Free PMC article. Review.
-
The molecular principles governing the activity and functional diversity of AAA+ proteins.Nat Rev Mol Cell Biol. 2020 Jan;21(1):43-58. doi: 10.1038/s41580-019-0183-6. Epub 2019 Nov 21. Nat Rev Mol Cell Biol. 2020. PMID: 31754261 Free PMC article. Review.
References
-
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. - PMC - PubMed
- Adams, P.D., Afonine, P.V., Bunkoczi, G., Chen, V.B., Davis, I.W., Echols, N., Headd, J.J., Hung, L.W., Kapral, G.J., Grosse-Kunstleve, R.W., et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213-221. - PMC - PubMed
-
- Biter A.B., Lee J., Sung N., Tsai F.T., Lee S. Functional analysis of conserved cis- and trans-elements in the Hsp104 protein disaggregating machine. J. Struct. Biol. 2012;179:172–180. - PMC - PubMed
- Biter, A.B., Lee, J., Sung, N., Tsai, F.T., and Lee, S. (2012). Functional analysis of conserved cis- and trans-elements in the Hsp104 protein disaggregating machine. J. Struct. Biol. 179, 172-180. - PMC - PubMed
-
- Carroni M., Kummer E., Oguchi Y., Wendler P., Clare D.K., Sinning I., Kopp J., Mogk A., Bukau B., Saibil H.R. Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. eLife. 2014;3:e02481. - PMC - PubMed
- Carroni, M., Kummer, E., Oguchi, Y., Wendler, P., Clare, D.K., Sinning, I., Kopp, J., Mogk, A., Bukau, B., and Saibil, H.R. (2014). Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. eLife 3, e02481. - PMC - PubMed
-
- Davies B.A., Azmi I.F., Payne J., Shestakova A., Horazdovsky B.F., Babst M., Katzmann D.J. Coordination of substrate binding and ATP hydrolysis in Vps4-mediated ESCRT-III disassembly. Mol. Biol. Cell. 2010;21:3396–3408. - PMC - PubMed
- Davies, B.A., Azmi, I.F., Payne, J., Shestakova, A., Horazdovsky, B.F., Babst, M., and Katzmann, D.J. (2010). Coordination of substrate binding and ATP hydrolysis in Vps4-mediated ESCRT-III disassembly. Mol. Biol. Cell 21, 3396-3408. - PMC - PubMed
-
- Davies B.A., Norgan A.P., Payne J.A., Schulz M.E., Nichols M.D., Tan J.A., Xu Z., Katzmann D.J. Vps4 stimulatory element of the cofactor Vta1 contacts the ATPase Vps4 α7 and α9 to stimulate ATP hydrolysis. J. Biol. Chem. 2014;289:28707–28718. - PMC - PubMed
- Davies, B.A., Norgan, A.P., Payne, J.A., Schulz, M.E., Nichols, M.D., Tan, J.A., Xu, Z., and Katzmann, D.J. (2014). Vps4 stimulatory element of the cofactor Vta1 contacts the ATPase Vps4 α7 and α9 to stimulate ATP hydrolysis. J. Biol. Chem. 289, 28707-28718. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
