

Severe influenza pneumonitis in children with inherited TLR3 deficiency
- PMID: 31217193
- PMCID: PMC6719423
- DOI: 10.1084/jem.20181621
Severe influenza pneumonitis in children with inherited TLR3 deficiency
Abstract
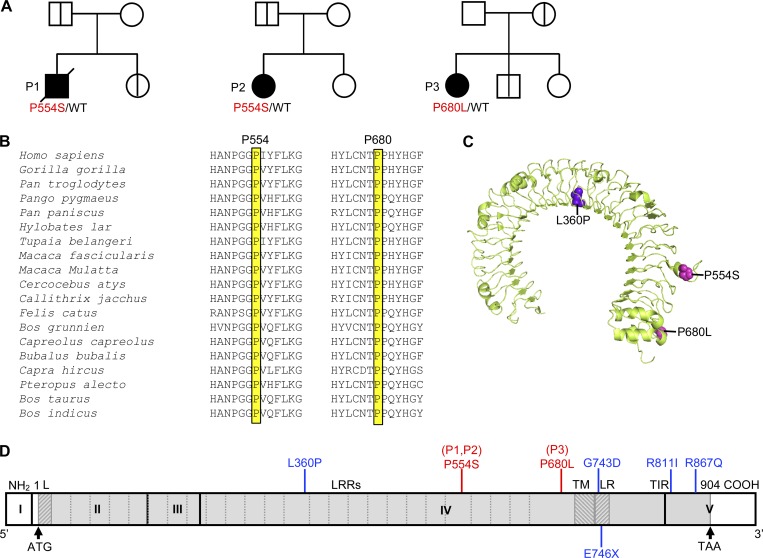
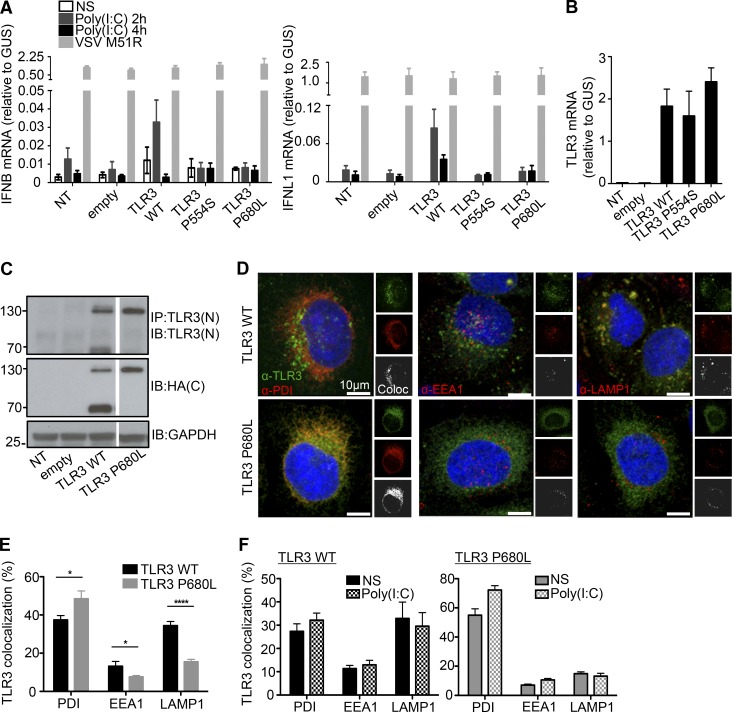
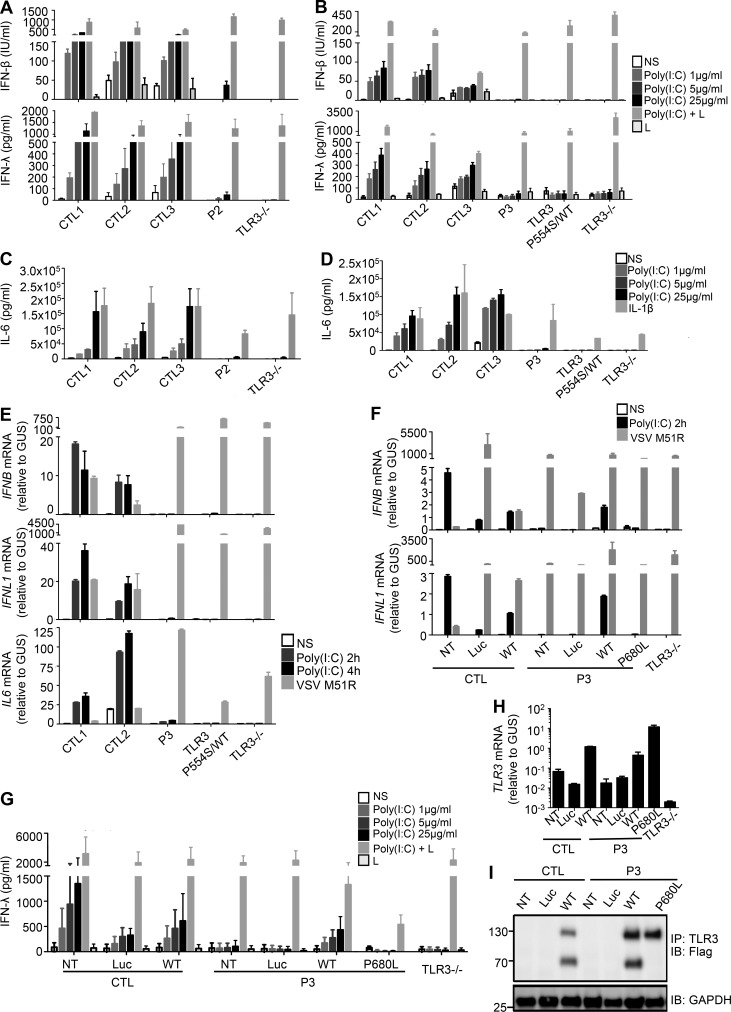
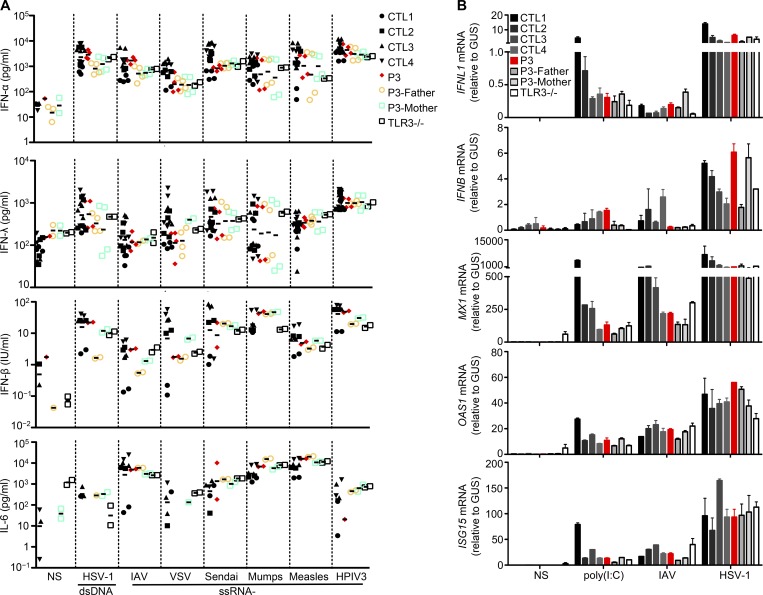
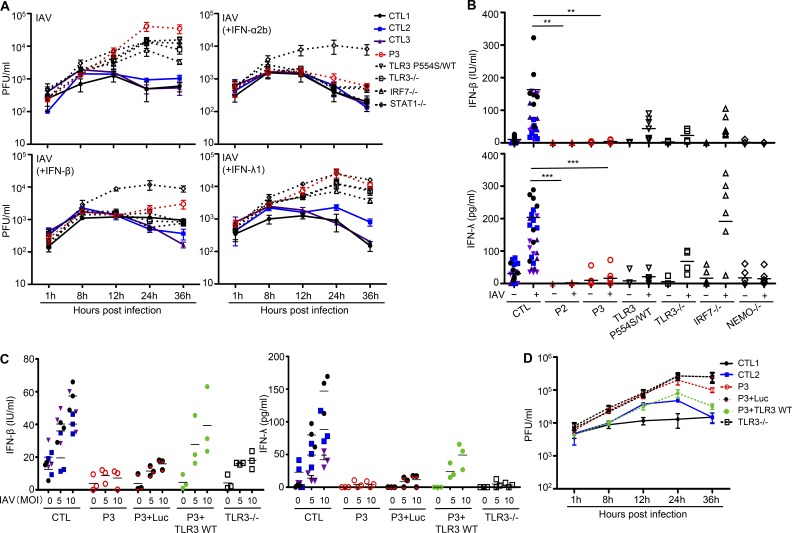
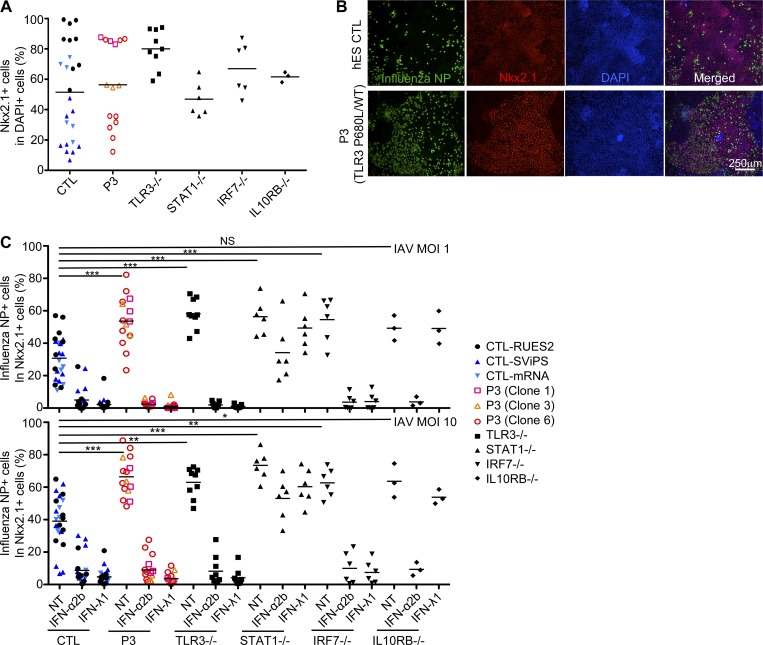
Autosomal recessive IRF7 and IRF9 deficiencies impair type I and III IFN immunity and underlie severe influenza pneumonitis. We report three unrelated children with influenza A virus (IAV) infection manifesting as acute respiratory distress syndrome (IAV-ARDS), heterozygous for rare TLR3 variants (P554S in two patients and P680L in the third) causing autosomal dominant (AD) TLR3 deficiency. AD TLR3 deficiency can underlie herpes simplex virus-1 (HSV-1) encephalitis (HSE) by impairing cortical neuron-intrinsic type I IFN immunity to HSV-1. TLR3-mutated leukocytes produce normal levels of IFNs in response to IAV. In contrast, TLR3-mutated fibroblasts produce lower levels of IFN-β and -λ, and display enhanced viral susceptibility, upon IAV infection. Moreover, the patients' iPSC-derived pulmonary epithelial cells (PECs) are susceptible to IAV. Treatment with IFN-α2b or IFN-λ1 rescues this phenotype. AD TLR3 deficiency may thus underlie IAV-ARDS by impairing TLR3-dependent, type I and/or III IFN-mediated, PEC-intrinsic immunity. Its clinical penetrance is incomplete for both IAV-ARDS and HSE, consistent with their typically sporadic nature.
© 2019 Lim et al.
Figures
References
-
- Ank N., Iversen M.B., Bartholdy C., Staeheli P., Hartmann R., Jensen U.B., Dagnaes-Hansen F., Thomsen A.R., Chen Z., Haugen H., et al. . 2008. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J. Immunol. 180:2474–2485. 10.4049/jimmunol.180.4.2474 - DOI - PubMed
-
- Audry M., Ciancanelli M., Yang K., Cobat A., Chang H.H., Sancho-Shimizu V., Lorenzo L., Niehues T., Reichenbach J., Li X.X., et al. . 2011. NEMO is a key component of NF-κB- and IRF-3-dependent TLR3-mediated immunity to herpes simplex virus. J. Allergy Clin. Immunol. 128:610–7.e1: 4. 10.1016/j.jaci.2011.04.059 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
