

Current best practices in single-cell RNA-seq analysis: a tutorial
- PMID: 31217225
- PMCID: PMC6582955
- DOI: 10.15252/msb.20188746
Current best practices in single-cell RNA-seq analysis: a tutorial
Abstract
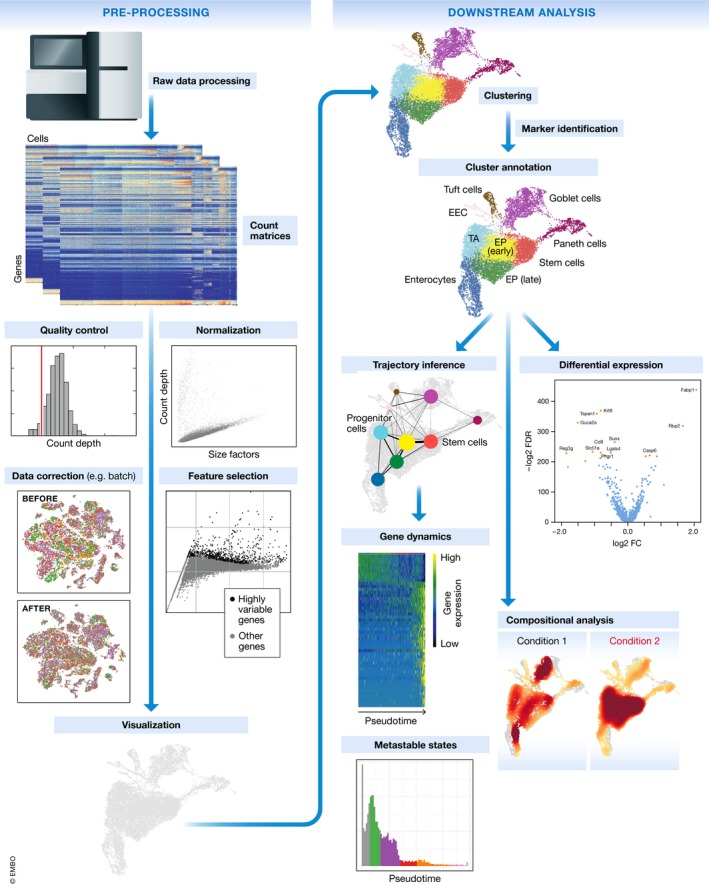
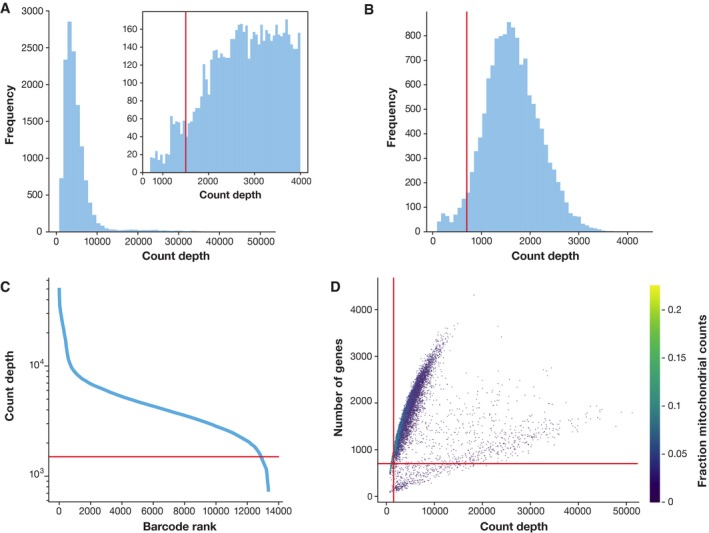
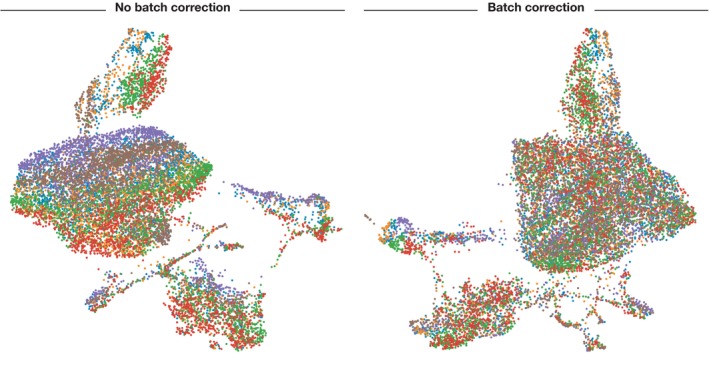
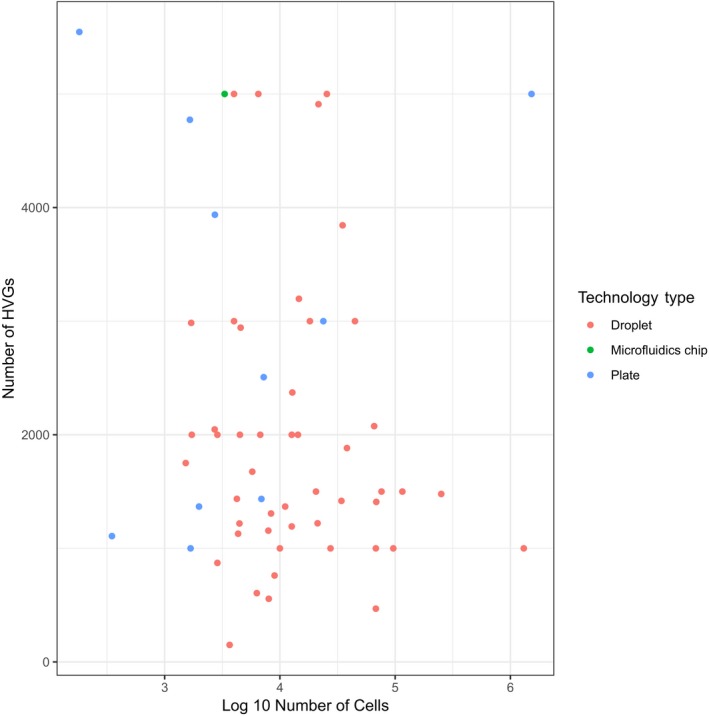
Single-cell RNA-seq has enabled gene expression to be studied at an unprecedented resolution. The promise of this technology is attracting a growing user base for single-cell analysis methods. As more analysis tools are becoming available, it is becoming increasingly difficult to navigate this landscape and produce an up-to-date workflow to analyse one's data. Here, we detail the steps of a typical single-cell RNA-seq analysis, including pre-processing (quality control, normalization, data correction, feature selection, and dimensionality reduction) and cell- and gene-level downstream analysis. We formulate current best-practice recommendations for these steps based on independent comparison studies. We have integrated these best-practice recommendations into a workflow, which we apply to a public dataset to further illustrate how these steps work in practice. Our documented case study can be found at https://www.github.com/theislab/single-cell-tutorial This review will serve as a workflow tutorial for new entrants into the field, and help established users update their analysis pipelines.
Keywords: analysis pipeline development; computational biology; data analysis tutorial; single‐cell RNA‐seq.
© 2019 The Authors. Published under the terms of the CC BY 4.0 license.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
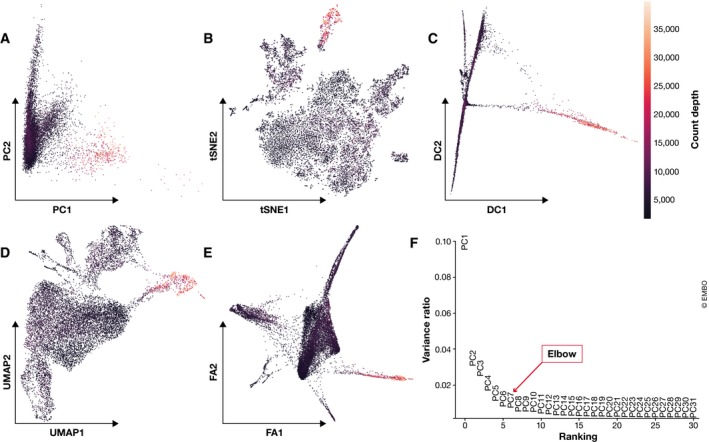
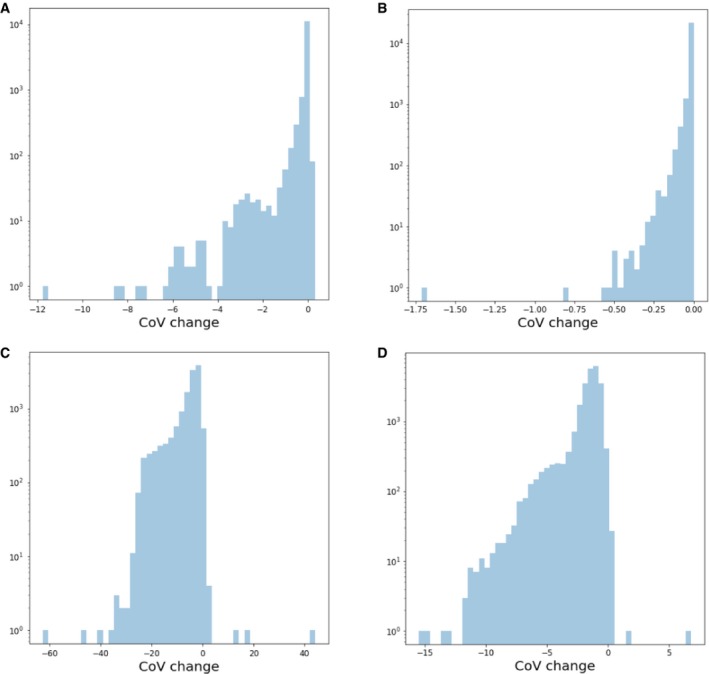
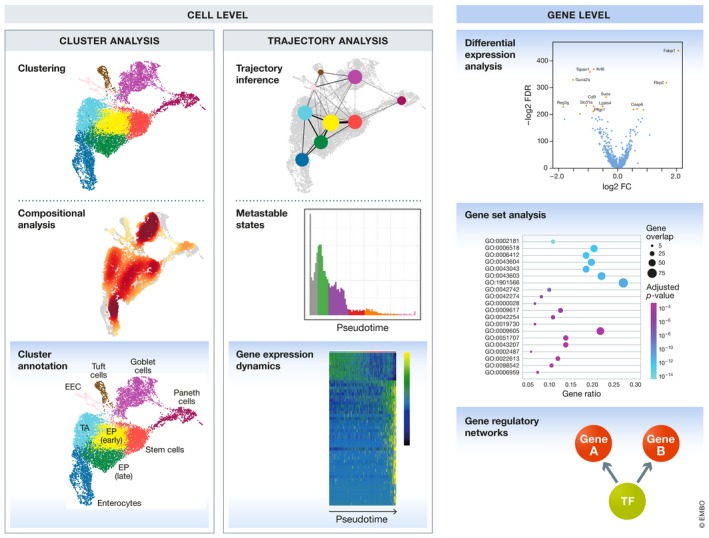
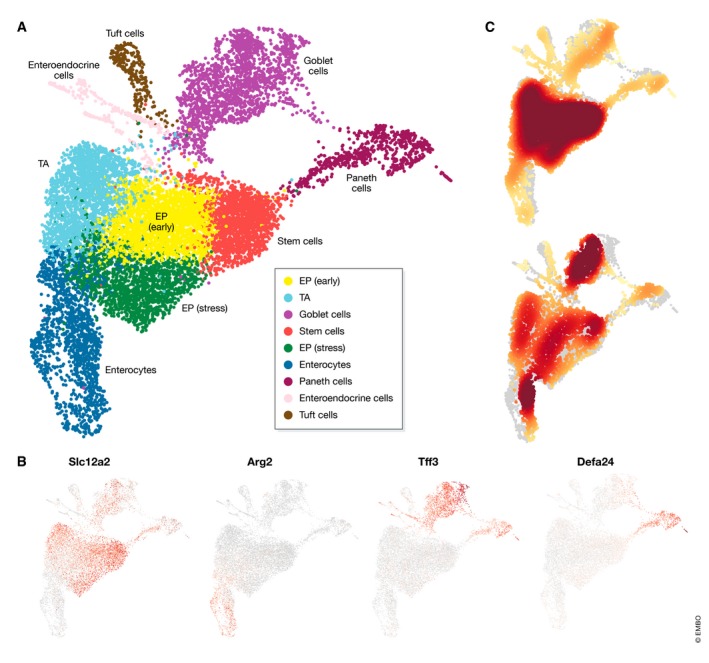
References
-
- Alpert A, Moore LS, Dubovik T, Shen‐Orr SS (2018) Alignment of single‐cell trajectories to compare cellular expression dynamics. Nat Methods 15: 267–270 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
