

PTEN Methylation by NSD2 Controls Cellular Sensitivity to DNA Damage
- PMID: 31217297
- PMCID: PMC6726527
- DOI: 10.1158/2159-8290.CD-18-0083
PTEN Methylation by NSD2 Controls Cellular Sensitivity to DNA Damage
Abstract
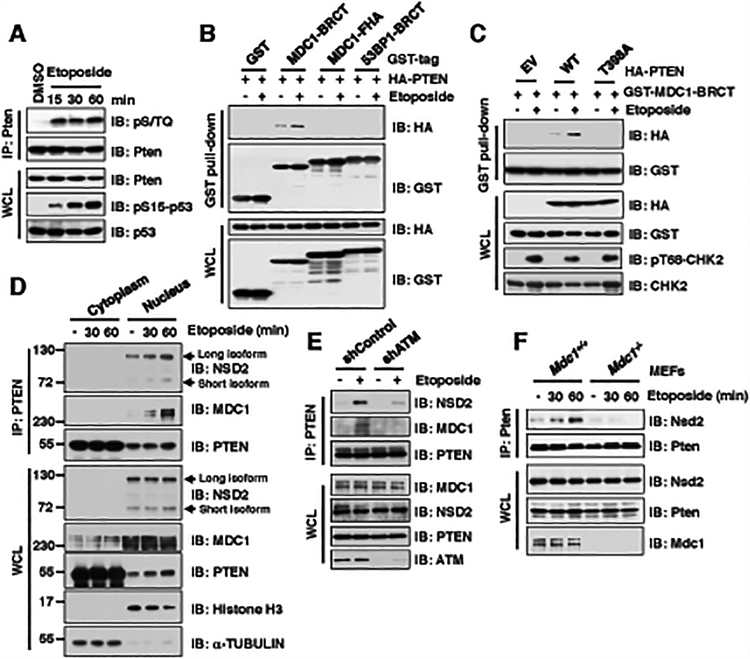
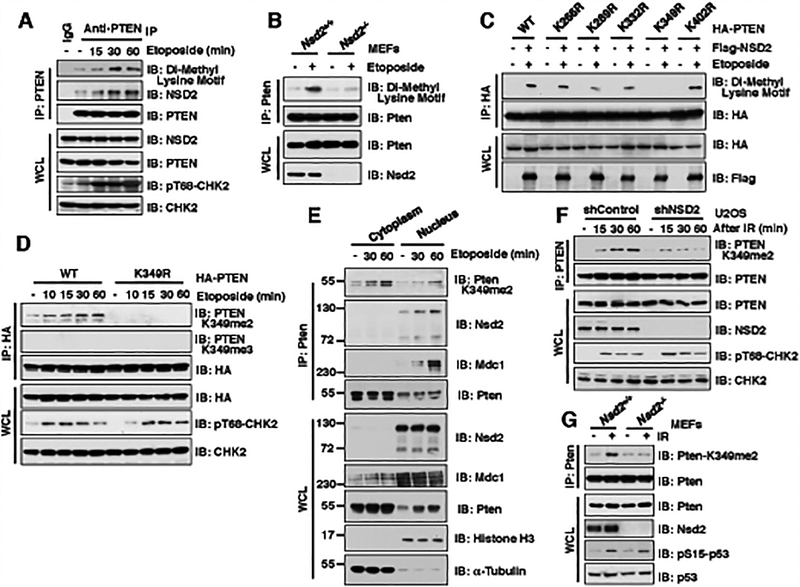
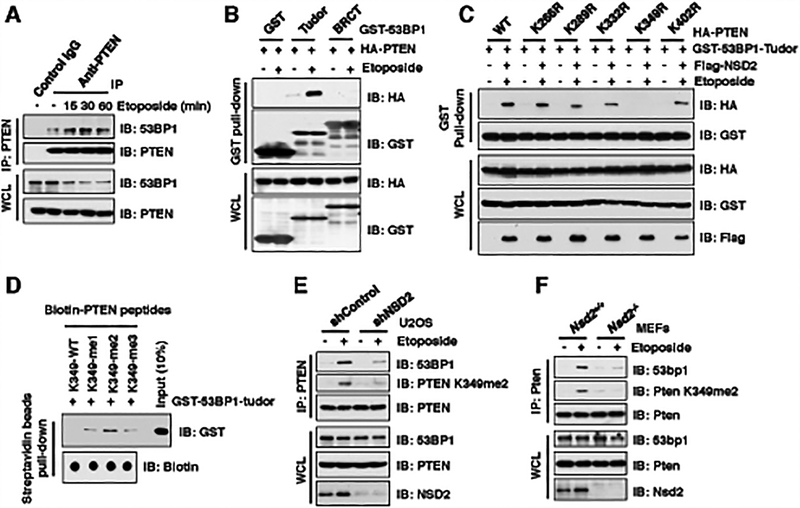
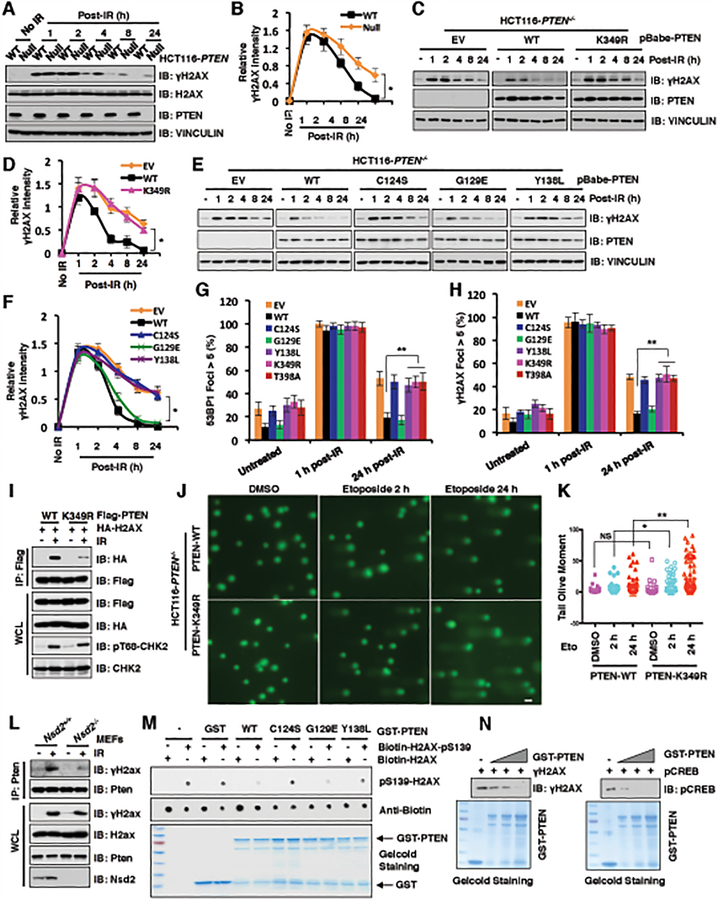
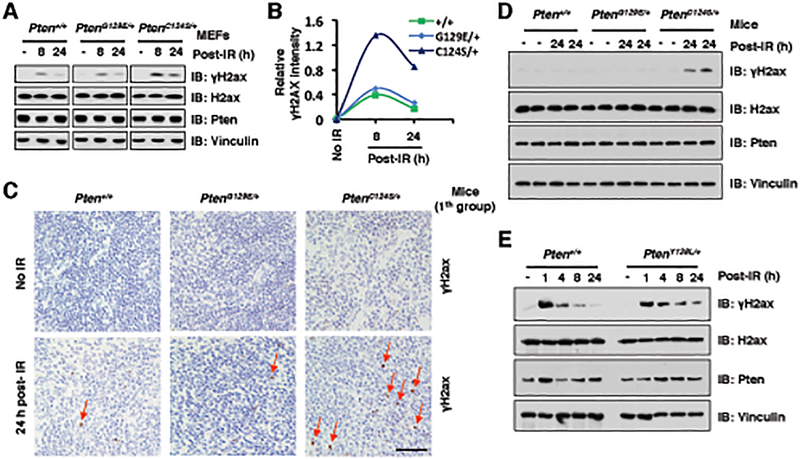
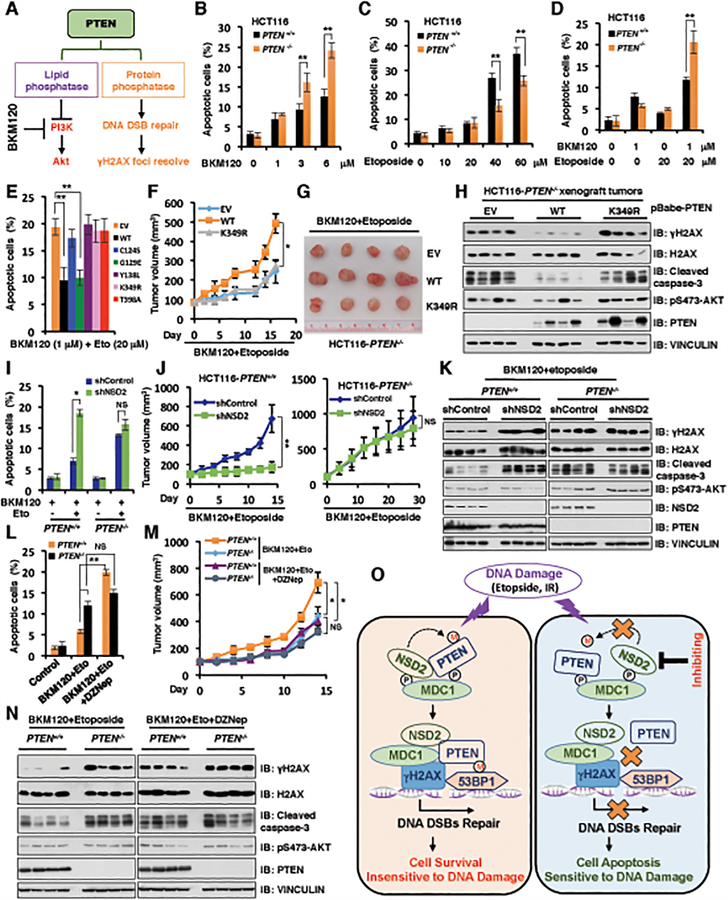
The function of PTEN in the cytoplasm largely depends on its lipid-phosphatase activity, though which it antagonizes the PI3K-AKT oncogenic pathway. However, molecular mechanisms underlying the role of PTEN in the nucleus remain largely elusive. Here, we report that DNA double-strand breaks (DSB) promote PTEN interaction with MDC1 upon ATM-dependent phosphorylation of T/S398-PTEN. Importantly, DNA DSBs enhance NSD2 (MMSET/WHSC1)-mediated dimethylation of PTEN at K349, which is recognized by the tudor domain of 53BP1 to recruit PTEN to DNA-damage sites, governing efficient repair of DSBs partly through dephosphorylation of γH2AX. Of note, inhibiting NSD2-mediated methylation of PTEN, either through expressing methylation-deficient PTEN mutants or through inhibiting NSD2, sensitizes cancer cells to combinatorial treatment with a PI3K inhibitor and DNA-damaging agents in both cell culture and in vivo xenograft models. Therefore, our study provides a novel molecular mechanism for PTEN regulation of DSB repair in a methylation- and protein phosphatase-dependent manner. SIGNIFICANCE: NSD2-mediated dimethylation of PTEN is recognized by the 53BP1 tudor domain to facilitate PTEN recruitment into DNA-damage sites, governing efficient repair of DNA DSBs. Importantly, inhibiting PTEN methylation sensitizes cancer cells to combinatorial treatment with a PI3K inhibitor combined with DNA-damaging agents in both cell culture and in vivo xenograft models.This article is highlighted in the In This Issue feature, p. 1143.
©2019 American Association for Cancer Research.
Conflict of interest statement
Figures
Similar articles
-
MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites.Nature. 2011 Feb 3;470(7332):124-8. doi: 10.1038/nature09658. Nature. 2011. PMID: 21293379 Free PMC article.
-
Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair.J Mol Cell Biol. 2013 Jun;5(3):157-65. doi: 10.1093/jmcb/mjs066. Epub 2013 Jan 16. J Mol Cell Biol. 2013. PMID: 23329852
-
Impact of histone H4 lysine 20 methylation on 53BP1 responses to chromosomal double strand breaks.PLoS One. 2012;7(11):e49211. doi: 10.1371/journal.pone.0049211. Epub 2012 Nov 28. PLoS One. 2012. PMID: 23209566 Free PMC article.
-
NSD2 as a Promising Target in Hematological Disorders.Int J Mol Sci. 2022 Sep 21;23(19):11075. doi: 10.3390/ijms231911075. Int J Mol Sci. 2022. PMID: 36232375 Free PMC article. Review.
-
The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax.DNA Repair (Amst). 2010 Dec 10;9(12):1273-82. doi: 10.1016/j.dnarep.2010.09.013. Epub 2010 Oct 30. DNA Repair (Amst). 2010. PMID: 21036673 Review.
Cited by
-
PTEN Nuclear Functions.Cold Spring Harb Perspect Med. 2020 May 1;10(5):a036079. doi: 10.1101/cshperspect.a036079. Cold Spring Harb Perspect Med. 2020. PMID: 31712221 Free PMC article. Review.
-
Drug Discovery Targeting Nuclear Receptor Binding SET Domain Protein 2 (NSD2).J Med Chem. 2023 Aug 24;66(16):10991-11026. doi: 10.1021/acs.jmedchem.3c00948. Epub 2023 Aug 14. J Med Chem. 2023. PMID: 37578463 Free PMC article. Review.
-
Targeting PTEN Regulation by Post Translational Modifications.Cancers (Basel). 2022 Nov 15;14(22):5613. doi: 10.3390/cancers14225613. Cancers (Basel). 2022. PMID: 36428706 Free PMC article. Review.
-
PTEN Tumor-Suppressor: The Dam of Stemness in Cancer.Cancers (Basel). 2019 Jul 30;11(8):1076. doi: 10.3390/cancers11081076. Cancers (Basel). 2019. PMID: 31366089 Free PMC article. Review.
-
Zinc finger protein 593 promotes breast cancer development by ensuring DNA damage repair and cell-cycle progression.iScience. 2024 Dec 3;27(12):111513. doi: 10.1016/j.isci.2024.111513. eCollection 2024 Dec 20. iScience. 2024. PMID: 39758980 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
