

A rare structural myopathy: Nemaline myopathy
- PMID: 31217710
- PMCID: PMC6559969
- DOI: 10.5152/TurkPediatriArs.2018.4402
A rare structural myopathy: Nemaline myopathy
Abstract
Nemaline myopathy, which is characterized by the accumulation of ''rod'' bodies in muscle fibers is a very rare inherited muscle disease. According to the underlying mutation, the disease has varying severity of clinical outcomes. Patients with severe forms of the disease die because of hypotonia, feeding difficulties, aspiration pneumonia, and respiratory failure in the neonatal or infancy period. Mild forms of the disease present with walking-swallowing difficulties and respiratory distress in late childhood or adulthood. A two-and-a-half-month-old boy was monitored in our Pediatric Intensive Care Unit with hypotonia, pneumonia, and respiratory distress. Nemaline myopathy was diagnosed as the result of a muscle biopsy. An advanced molecular examination revealed heterozygous mutations in the skeletal muscle α-actin (ACTA1) gene, which is the second most common cause of this disease. Nemaline myopathy should be kept in mind in patients of all age groups with respiratory failure and walking difficulty secondary to muscle weakness.
Nemalin miyopatisi oldukça nadir görülen kalıtımsal bir kas hastalığı olup kas liflerinde ‘’rod’’(nemalin) cisimciği birikimi ile tanımlanmaktadır. Hastalık altta yatan mutasyona ve mutasyonun kalıtım biçimine göre değişen ağırlıkta klinik gidişe sahiptir. Ağır şekillerinde olgular yutma ve solunum kaslarının etkilenmesi sonucu beslenme yetersizliği, aspirasyon pnömonisi ve solunum yetmezliği nedeni ile yenidoğan ya da süt çocukluğu döneminde kaybedilmektedir. Geç başlangıçlı hafif olgular yaşam kalitesini bozan yürüme-yutma zorluğu ve solunum sıkıntısı ile geç çocukluk ya da erişkin yaşta bulgu verebilmektedir. Hipotoni, pnömoni ve solunum sıkıntısı ile Çocuk Yoğun Bakım Birimi’nde izlenen iki buçuk aylık erkek bebeğe kas biyopsisi sonucu nemalin miyopatisi tanısı koyuldu. İleri moleküler inceleme sonucu hastalığın ikinci en sık nedeni olan “Skeletal Muscle α-Actin” (ACTA1) geninde heterozigot mutasyon saptandı. Yenidoğan döneminden erişkin döneme kadar kas güçsüzlüğüne bağlı solunum yetmezliği ve yutma-yürüme güçlüğü varlığında yapısal miyopatiler içinde nemalin miyopatisi akılda bulundurulmalı, şüphenilen olgulara kas biyopsisi ya/ya da genetik inceleme yapılmalıdır.
Keywords: ACTA1 gene; hypotonia; muscle biopsy; nemaline myopathy; respiratory failure.
Conflict of interest statement
Conflict of Interest: No conflict of interest was declared by the authors.
Figures
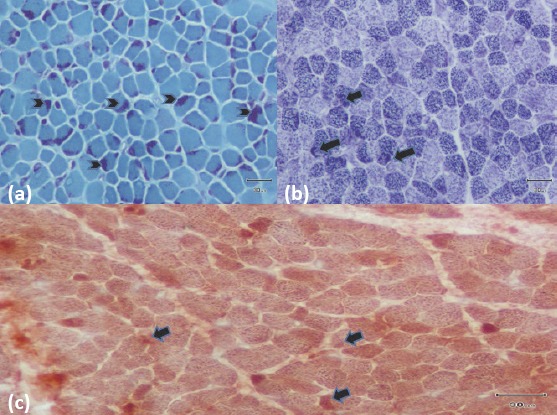
Similar articles
-
[Sibling cases of severe infantile form of nemaline myopathy with ACTA1-gene mutation].No To Hattatsu. 2013 Nov;45(6):452-6. No To Hattatsu. 2013. PMID: 24313005 Japanese.
-
Follow-up of nemaline myopathy in two patients with novel mutations in the skeletal muscle alpha-actin gene (ACTA1).Neuromuscul Disord. 2004 Sep;14(8-9):471-5. doi: 10.1016/j.nmd.2004.05.016. Neuromuscul Disord. 2004. PMID: 15336687
-
[Nemaline myopathy as a cause of neonatal hypotonia - with emphasis on personal experiences. Report of a family with two brothers affected].Med Wieku Rozwoj. 2009 Jan-Mar;13(1):5-10. Med Wieku Rozwoj. 2009. PMID: 19648653 Polish.
-
Clinical and Histologic Findings in ACTA1-Related Nemaline Myopathy: Case Series and Review of the Literature.Pediatr Neurol. 2017 Oct;75:11-16. doi: 10.1016/j.pediatrneurol.2017.04.002. Epub 2017 Apr 7. Pediatr Neurol. 2017. PMID: 28780987 Review.
-
Congenital myopathy with cap-like structures and nemaline rods: case report and literature review.Pediatr Neurol. 2014 Aug;51(2):192-7. doi: 10.1016/j.pediatrneurol.2014.04.002. Epub 2014 Apr 12. Pediatr Neurol. 2014. PMID: 25079567 Review.
Cited by
-
Nemaline myopathy with dilated cardiomyopathy and severe heart failure: A case report.World J Clin Cases. 2021 Apr 16;9(11):2569-2575. doi: 10.12998/wjcc.v9.i11.2569. World J Clin Cases. 2021. PMID: 33889622 Free PMC article.
References
-
- North KN, Ryan MM. In: Nemaline myopathy. GeneReviews. Pagon RA, Adam MP, Ardinger HH, editors. Seattle: University of Washington, Seattle; 2015. pp. 1993–2019. - PubMed
-
- Yin X, Pu CQ, Wang Q, Liu JX, Mao YL. Clinical and pathological features of patients with nemaline myopathy. Mol Med Rep. 2014;10:175–82. - PubMed
-
- Shy GM, Engel WK, Somers JE, Wanko T. Nemaline Myopathy. A New Congenital Myopathy. Brain. 1963;86:793–810. - PubMed
-
- Ryan MM, Schnell C, Strickland CD, et al. Nemaline myopathy:a clinical study of 143 cases. Ann Neurol. 2001;50:312–20. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
