

A decade of clinical development of PARP inhibitors in perspective
- PMID: 31218365
- PMCID: PMC6771225
- DOI: 10.1093/annonc/mdz192
A decade of clinical development of PARP inhibitors in perspective
Abstract
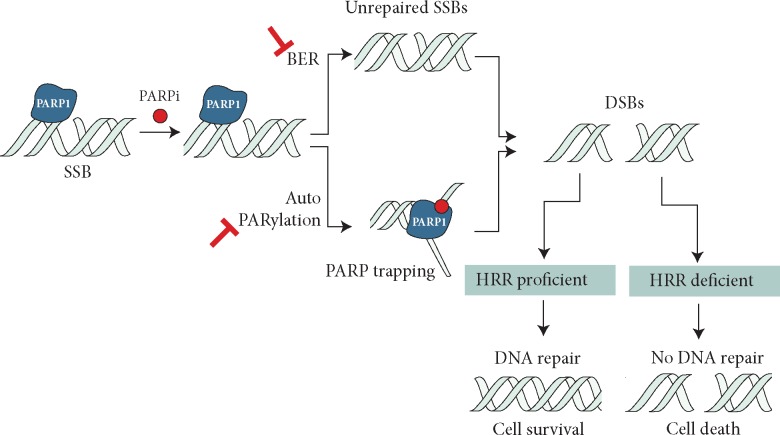
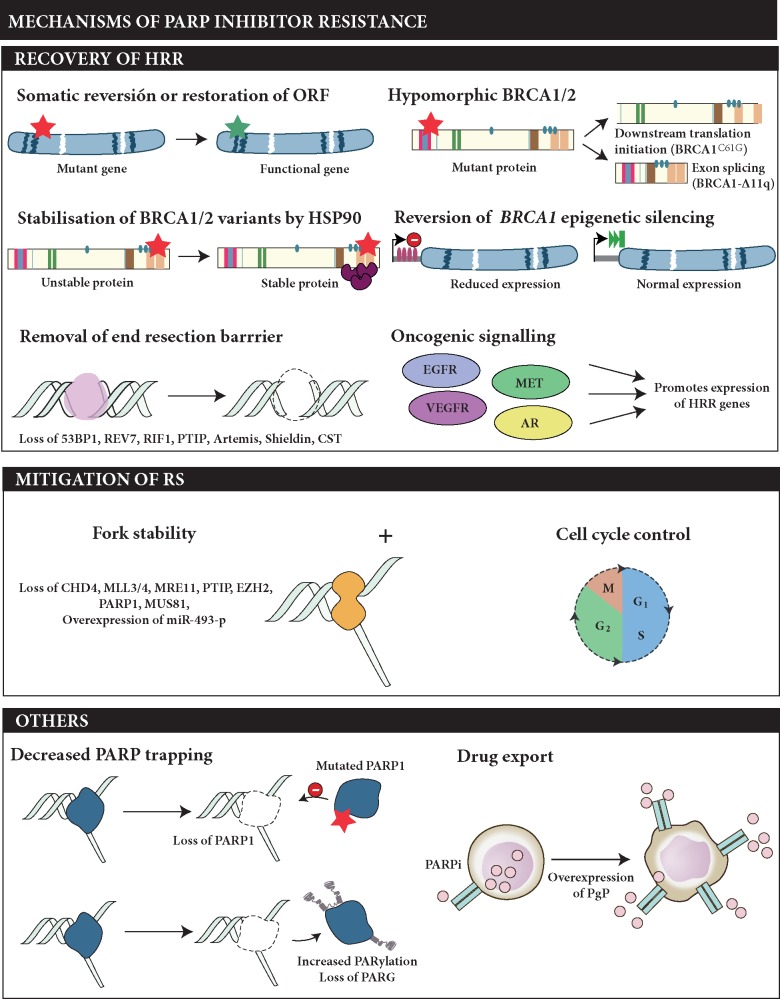
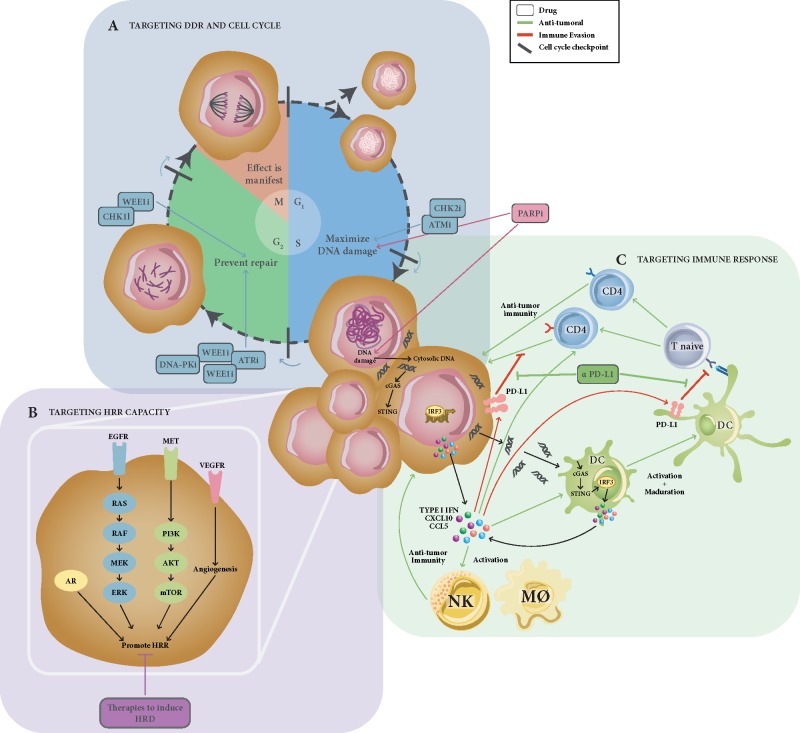
Genomic instability is a hallmark of cancer, and often is the result of altered DNA repair capacities in tumour cells. DNA damage repair defects are common in different cancer types; these alterations can also induce tumour-specific vulnerabilities that can be exploited therapeutically. In 2009, a first-in-man clinical trial of the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib clinically validated the synthetic lethal interaction between inhibition of PARP1, a key sensor of DNA damage, and BRCA1/BRCA2 deficiency. In this review, we summarize a decade of PARP inhibitor clinical development, a work that has resulted in the registration of several PARP inhibitors in breast (olaparib and talazoparib) and ovarian cancer (olaparib, niraparib and rucaparib, either alone or following platinum chemotherapy as maintenance therapy). Over the past 10 years, our knowledge on the mechanism of action of PARP inhibitor as well as how tumours become resistant has been extended, and we summarise this work here. We also discuss opportunities for expanding the precision medicine approach with PARP inhibitors, identifying a wider population who could benefit from this drug class. This includes developing and validating better predictive biomarkers for patient stratification, mainly based on homologous recombination defects beyond BRCA1/BRCA2 mutations, identifying DNA repair deficient tumours in other cancer types such as prostate or pancreatic cancer, or by designing combination therapies with PARP inhibitors.
Keywords: DNA repair; PARP inhibitors; clinical trials.
© The Author(s) 2019. Published by Oxford University Press on behalf of the European Society for Medical Oncology.
Figures
References
-
- Farmer H, McCabe N, Lord CJ. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434(7035): 917–921. - PubMed
-
- Bryant HE, Schultz N, Thomas HD. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434(7035): 913–917. - PubMed
-
- McCabe N, Turner NC, Lord CJ. et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 2006; 66(16): 8109–8115. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
