

Comment on 'Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size'
- PMID: 31219782
- PMCID: PMC6586461
- DOI: 10.7554/eLife.44718
Comment on 'Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size'
Abstract
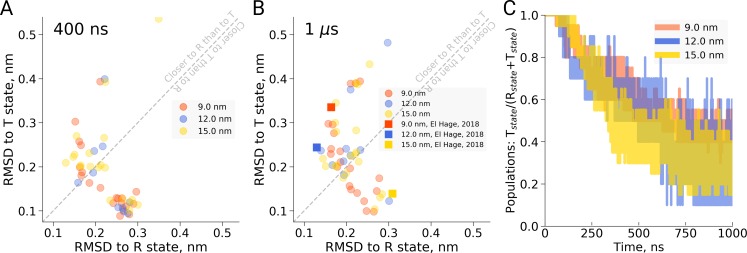
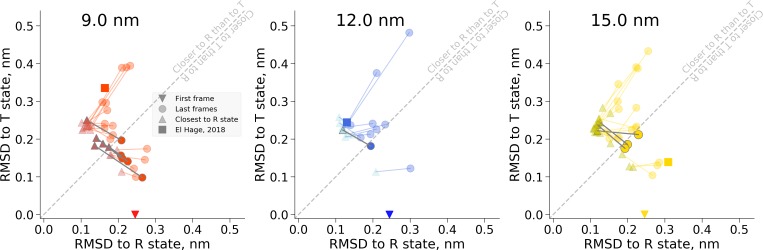
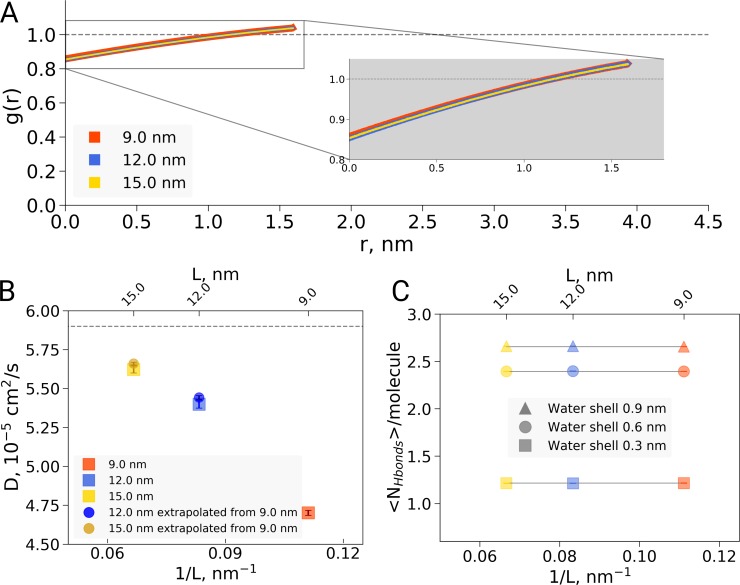
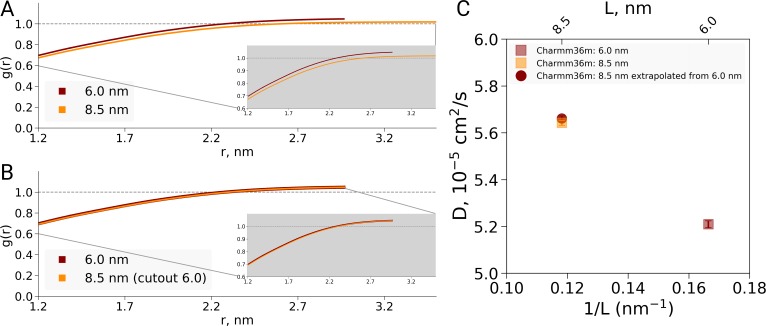
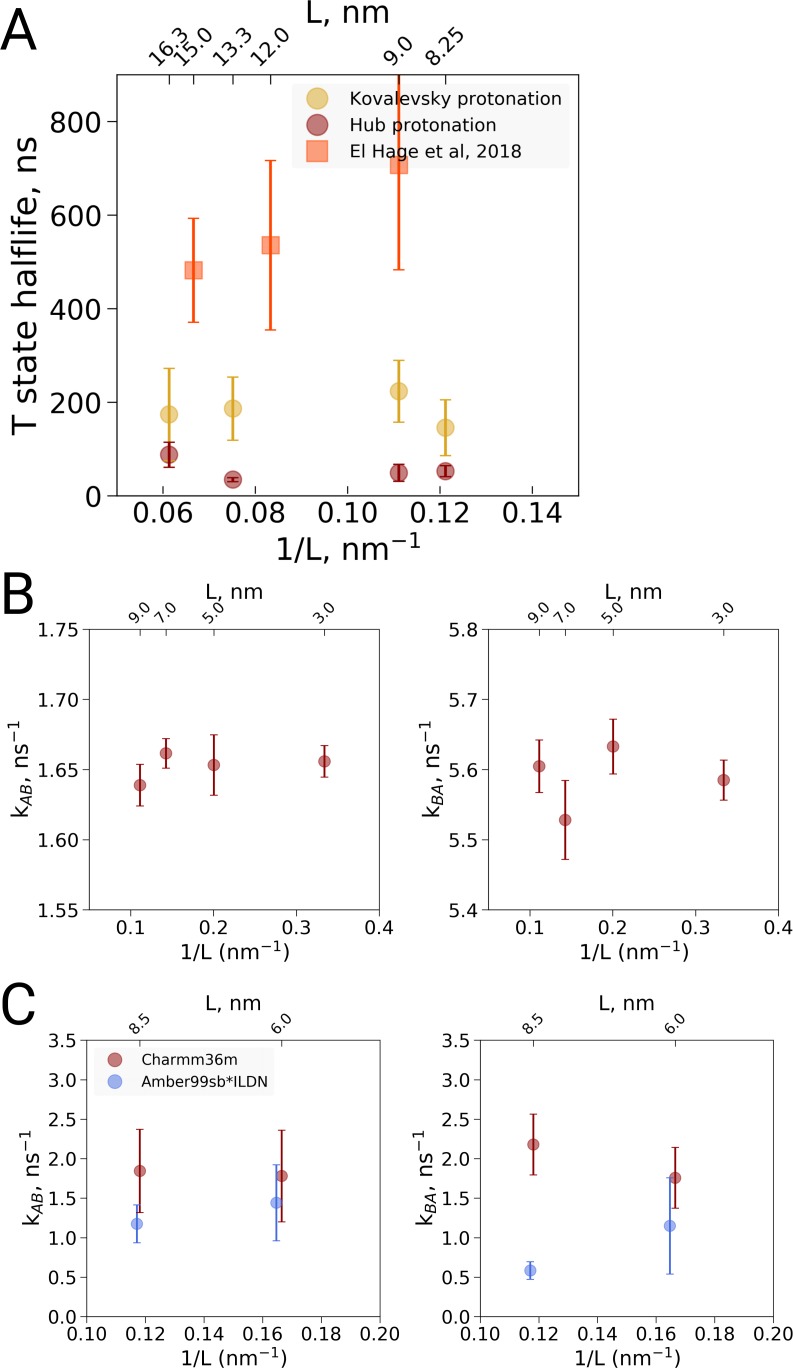
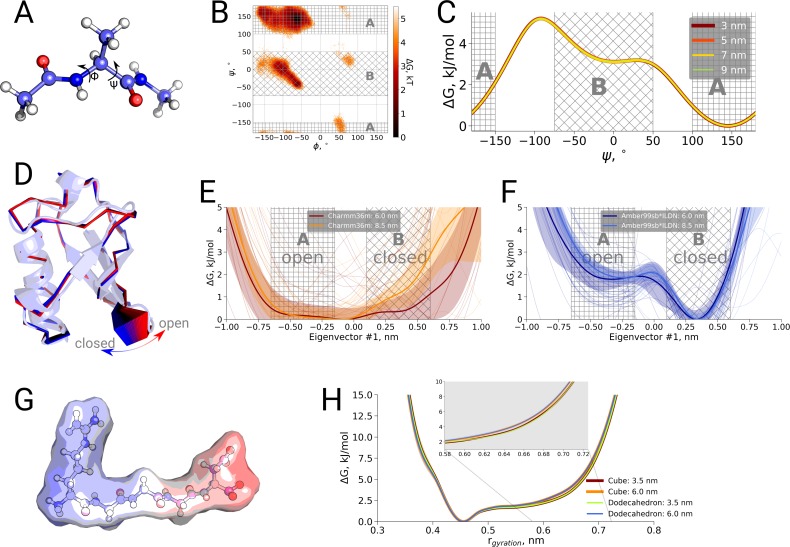
A recent molecular dynamics investigation into the stability of hemoglobin concluded that the unliganded protein is only stable in the T state when a solvent box is used in the simulations that is ten times larger than what is usually employed (El Hage et al., 2018). Here, we express three main concerns about that study. In addition, we find that with an order of magnitude more statistics, the reported box size dependence is not reproducible. Overall, no significant effects on the kinetics or thermodynamics of conformational transitions were observed.
Keywords: box size; hemoglobin; human; kinetics; molecular biophysics; molecular dynamics; structural biology; thermodynamics.
© 2019, Gapsys and de Groot.
Conflict of interest statement
VG, Bd No competing interests declared
Figures
Comment in
-
Response to comment on 'Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size'.Elife. 2019 Jun 20;8:e45318. doi: 10.7554/eLife.45318. Elife. 2019. PMID: 31219783 Free PMC article.
Comment on
-
Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size.Elife. 2018 Jul 12;7:e35560. doi: 10.7554/eLife.35560. Elife. 2018. PMID: 29998846 Free PMC article.
References
-
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. - DOI
-
- Darden T, York D, Pedersen L. Particle mesh Ewald: An N ⋅log( N ) method for Ewald sums in large systems. Journal of Chemical Physics. 1993;98:10089–10092. doi: 10.1063/1.464397. - DOI
-
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. Journal of Chemical Physics. 1995;103:8577–8593. doi: 10.1063/1.470117. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
