

Deficiency of nucleotide excision repair is associated with mutational signature observed in cancer
- PMID: 31221724
- PMCID: PMC6633256
- DOI: 10.1101/gr.246223.118
Deficiency of nucleotide excision repair is associated with mutational signature observed in cancer
Abstract
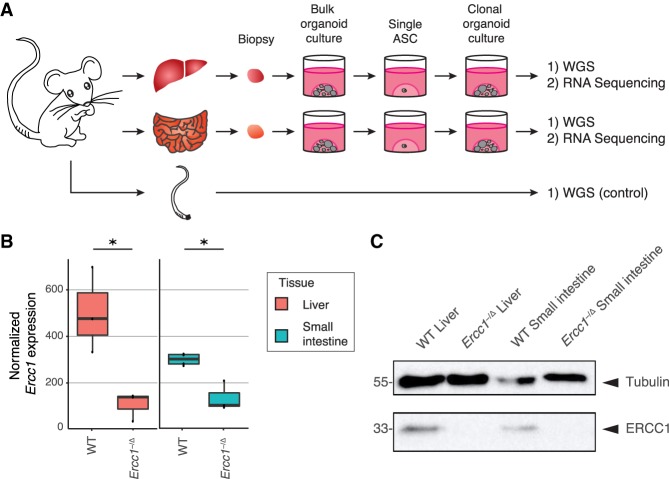
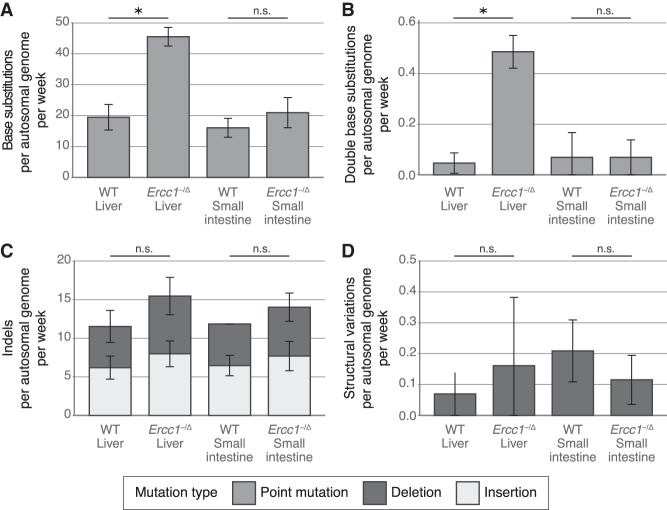
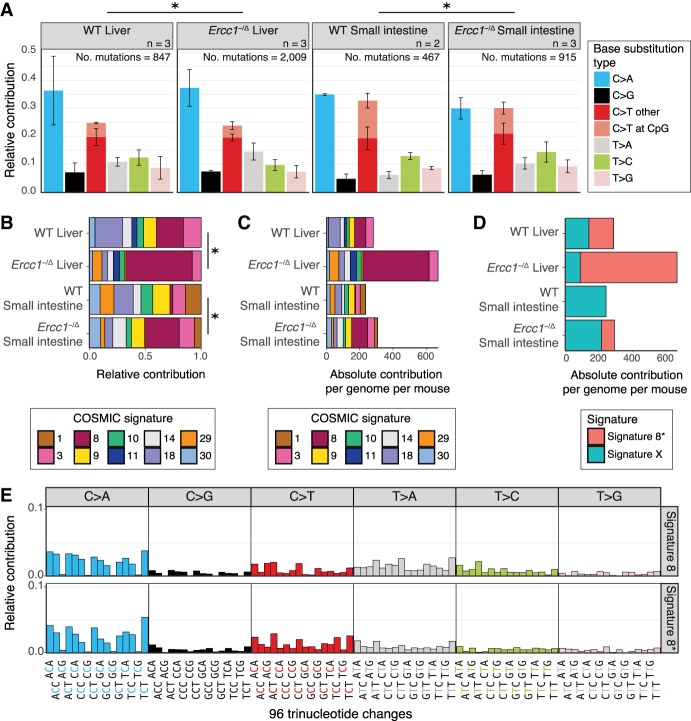
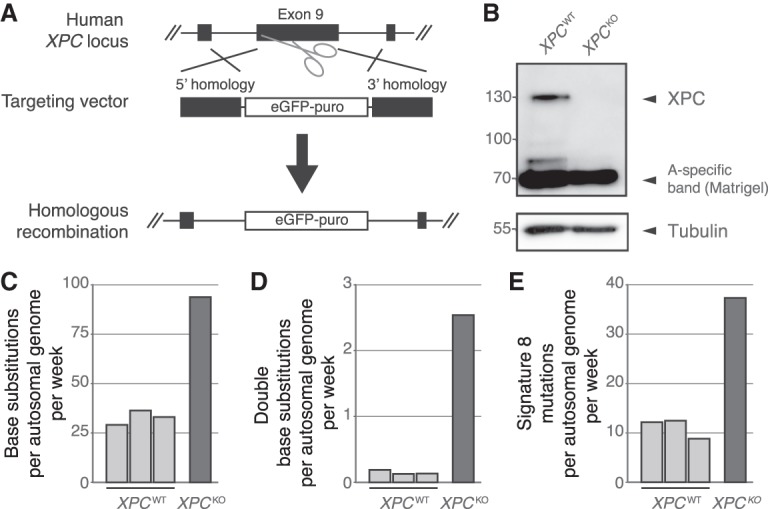
Nucleotide excision repair (NER) is one of the main DNA repair pathways that protect cells against genomic damage. Disruption of this pathway can contribute to the development of cancer and accelerate aging. Mutational characteristics of NER-deficiency may reveal important diagnostic opportunities, as tumors deficient in NER are more sensitive to certain treatments. Here, we analyzed the genome-wide somatic mutational profiles of adult stem cells (ASCs) from NER-deficient Ercc1 -/Δ mice. Our results indicate that NER-deficiency increases the base substitution load twofold in liver but not in small intestinal ASCs, which coincides with the tissue-specific aging pathology observed in these mice. Moreover, NER-deficient ASCs of both tissues show an increased contribution of Signature 8 mutations, which is a mutational pattern with unknown etiology that is recurrently observed in various cancer types. The scattered genomic distribution of the base substitutions indicates that deficiency of global-genome NER (GG-NER) underlies the observed mutational consequences. In line with this, we observe increased Signature 8 mutations in a GG-NER-deficient human organoid culture, in which XPC was deleted using CRISPR-Cas9 gene-editing. Furthermore, genomes of NER-deficient breast tumors show an increased contribution of Signature 8 mutations compared with NER-proficient tumors. Elevated levels of Signature 8 mutations could therefore contribute to a predictor of NER-deficiency based on a patient's mutational profile.
© 2019 Jager et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Alexandrov L, Kim J, Haradhvala NJ, Huang MN, Ng AWT, Boot A, Covington KR, Gordenin DA, Bergstrom E, Lopez-Bigas N, et al. 2018. The repertoire of mutational signatures in human cancer. bioRxiv 10.1101/322859 - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources