

A cell topography-based mechanism for ligand discrimination by the T cell receptor
- PMID: 31221762
- PMCID: PMC6628812
- DOI: 10.1073/pnas.1817255116
A cell topography-based mechanism for ligand discrimination by the T cell receptor
Abstract
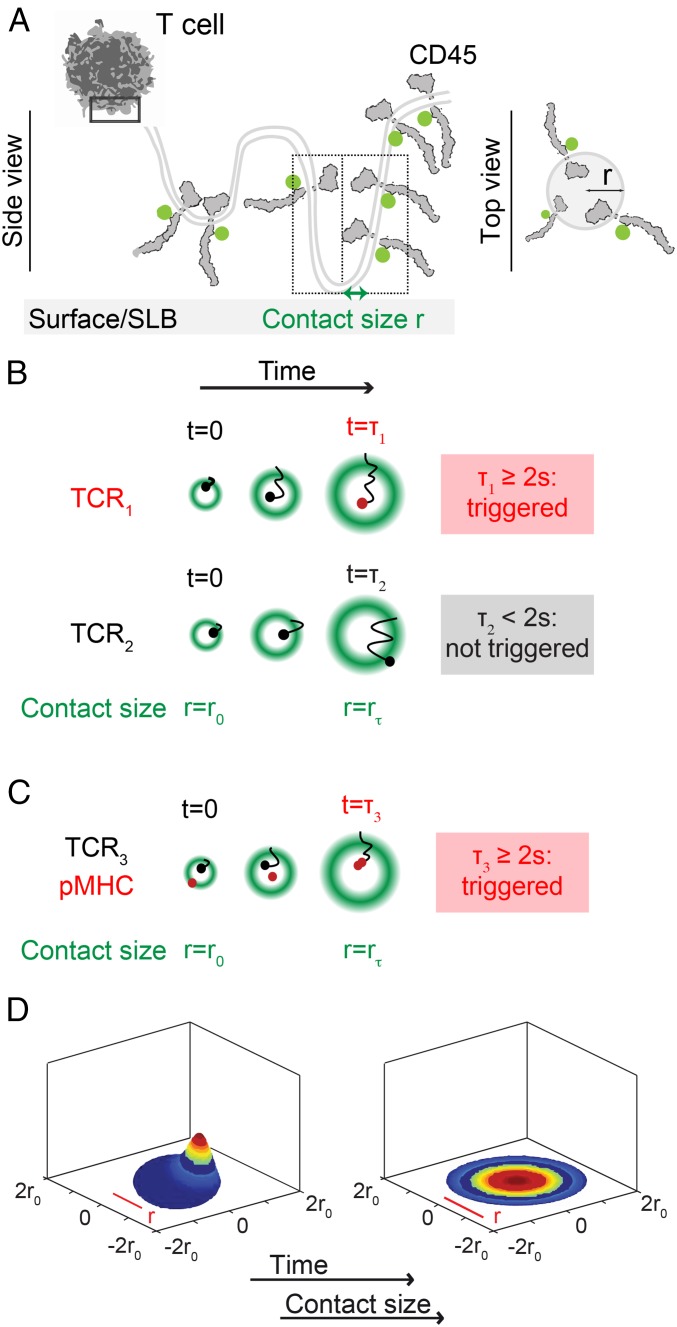
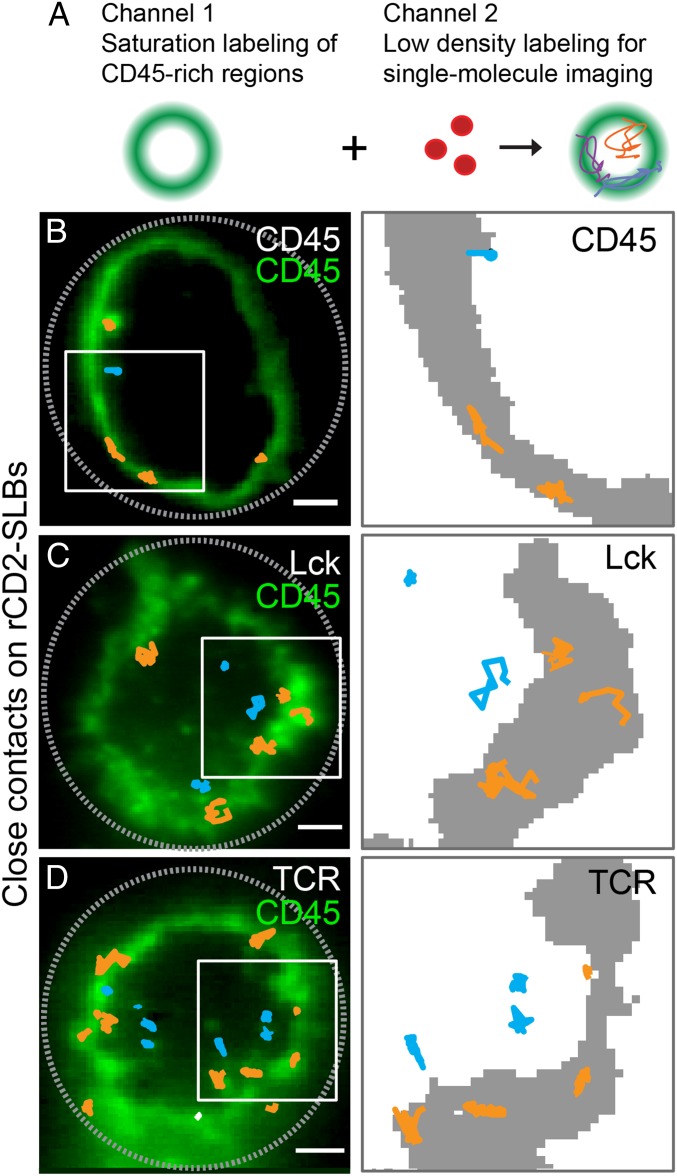
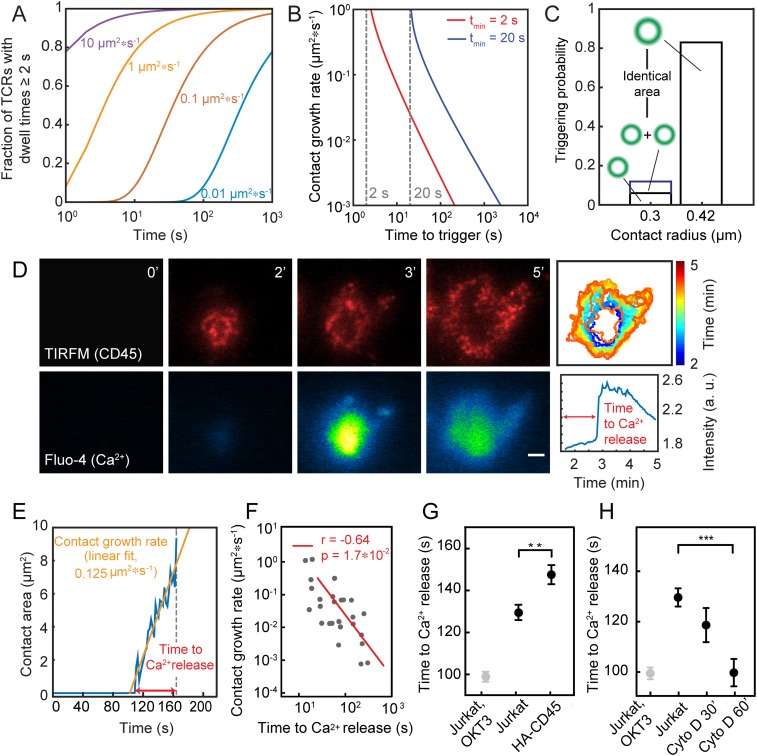
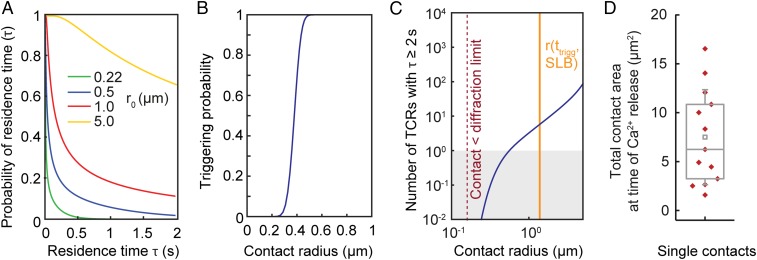
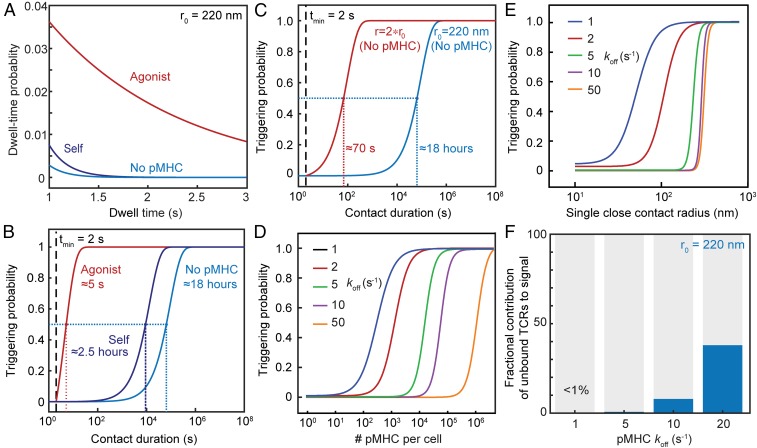
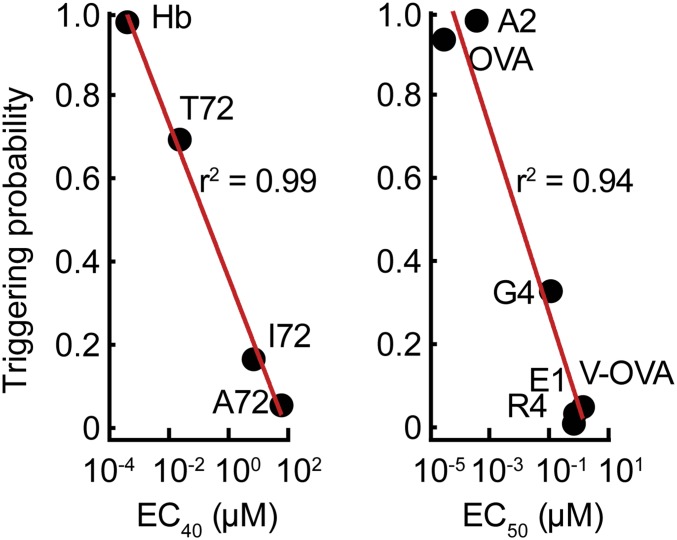
The T cell receptor (TCR) initiates the elimination of pathogens and tumors by T cells. To avoid damage to the host, the receptor must be capable of discriminating between wild-type and mutated self and nonself peptide ligands presented by host cells. Exactly how the TCR does this is unknown. In resting T cells, the TCR is largely unphosphorylated due to the dominance of phosphatases over the kinases expressed at the cell surface. However, when agonist peptides are presented to the TCR by major histocompatibility complex proteins expressed by antigen-presenting cells (APCs), very fast receptor triggering, i.e., TCR phosphorylation, occurs. Recent work suggests that this depends on the local exclusion of the phosphatases from regions of contact of the T cells with the APCs. Here, we developed and tested a quantitative treatment of receptor triggering reliant only on TCR dwell time in phosphatase-depleted cell contacts constrained in area by cell topography. Using the model and experimentally derived parameters, we found that ligand discrimination likely depends crucially on individual contacts being ∼200 nm in radius, matching the dimensions of the surface protrusions used by T cells to interrogate their targets. The model not only correctly predicted the relative signaling potencies of known agonists and nonagonists but also achieved this in the absence of kinetic proofreading. Our work provides a simple, quantitative, and predictive molecular framework for understanding why TCR triggering is so selective and fast and reveals that, for some receptors, cell topography likely influences signaling outcomes.
Keywords: T cell receptor; dwell time; microvilli; receptor triggering; single-molecule imaging.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Morris G. P., Allen P. M., How the TCR balances sensitivity and specificity for the recognition of self and pathogens. Nat. Immunol. 13, 121–128 (2012). - PubMed
-
- Wong H. S., Germain R. N., Robust control of the adaptive immune system. Semin. Immunol. 36, 17–27 (2018). - PubMed
-
- Kershaw M. H., Westwood J. A., Darcy P. K., Gene-engineered T cells for cancer therapy. Nat. Rev. Cancer 13, 525–541 (2013). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
