

Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia
- PMID: 31222014
- PMCID: PMC6586637
- DOI: 10.1038/s41467-019-10652-9
Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia
Abstract
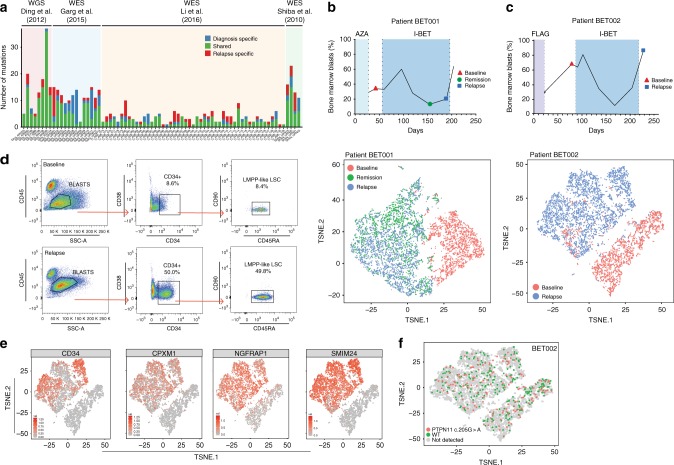
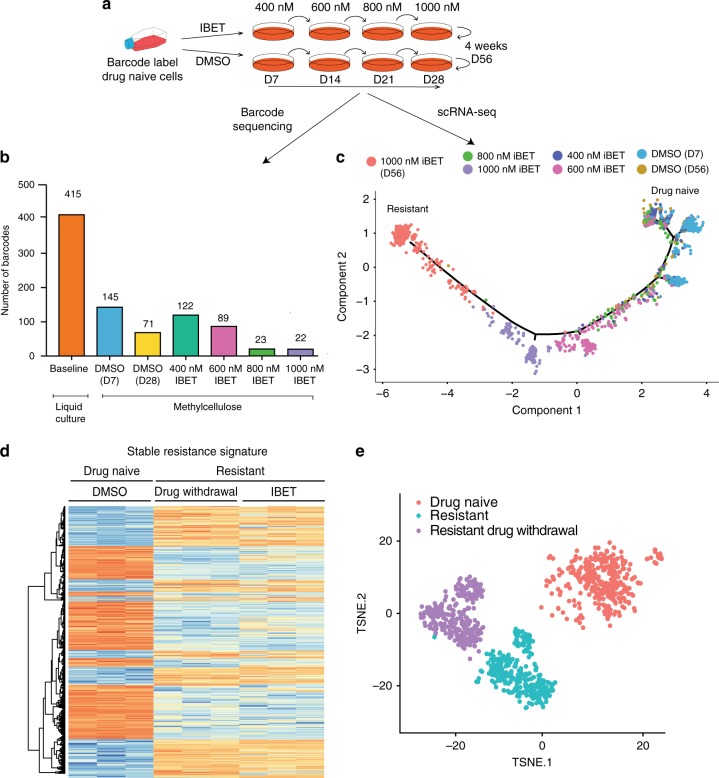
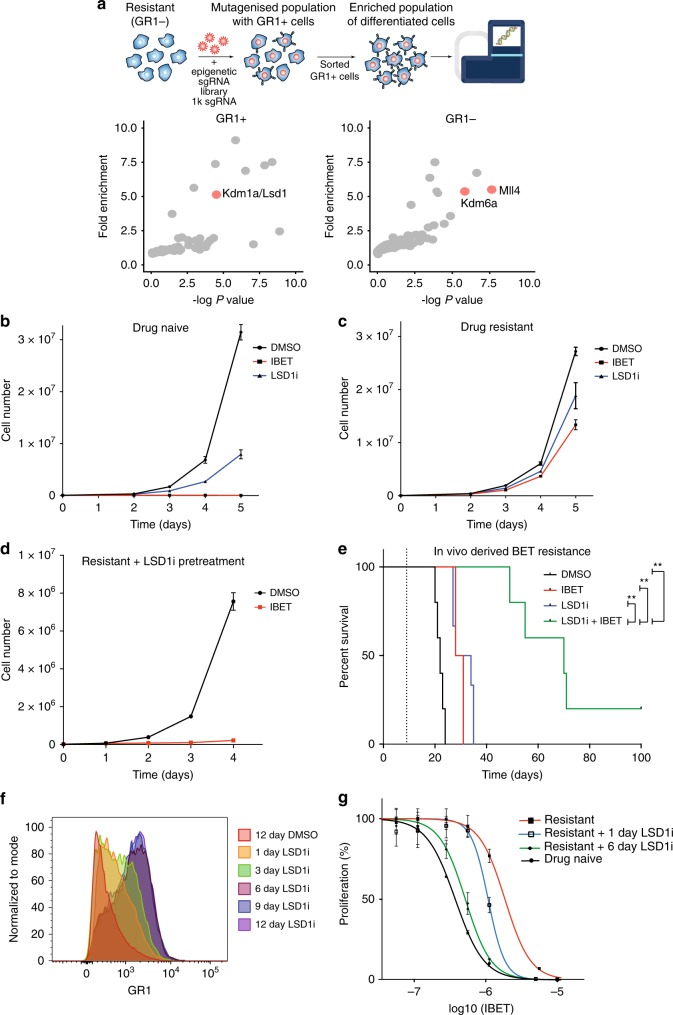
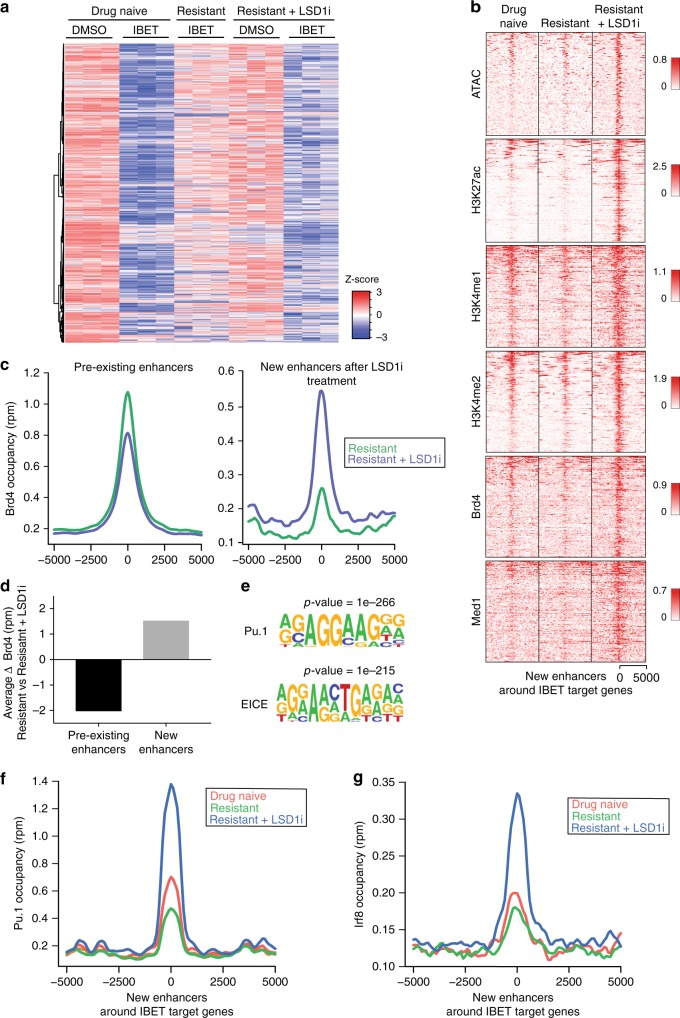
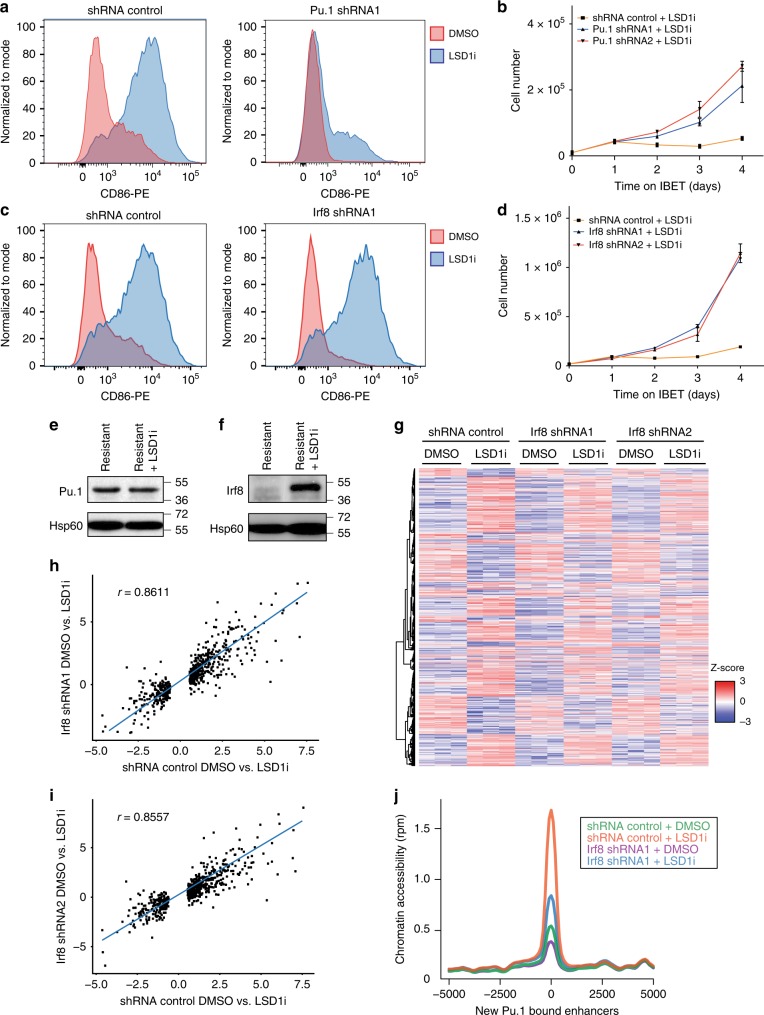
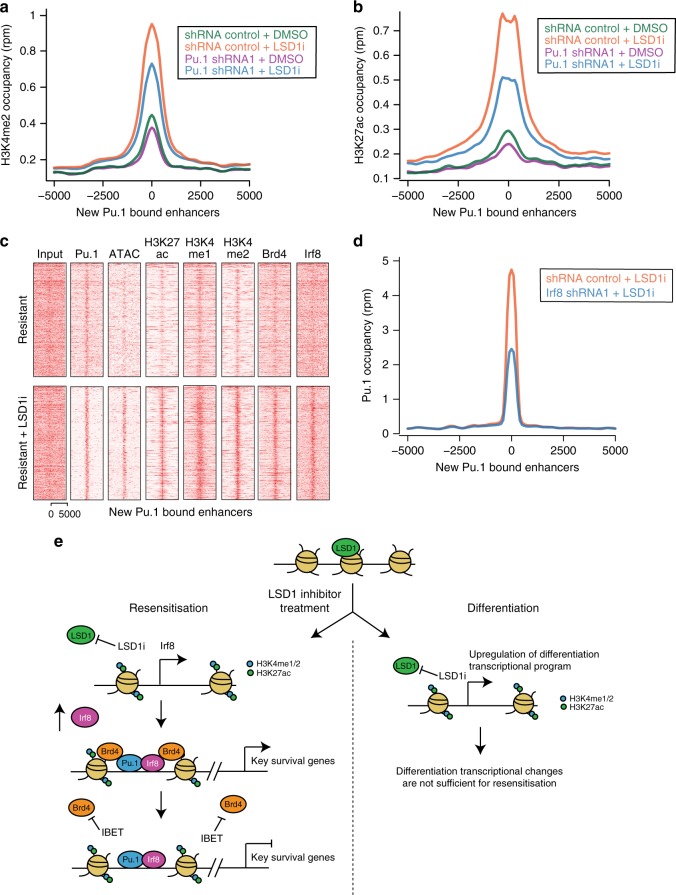
Non-genetic drug resistance is increasingly recognised in various cancers. Molecular insights into this process are lacking and it is unknown whether stable non-genetic resistance can be overcome. Using single cell RNA-sequencing of paired drug naïve and resistant AML patient samples and cellular barcoding in a unique mouse model of non-genetic resistance, here we demonstrate that transcriptional plasticity drives stable epigenetic resistance. With a CRISPR-Cas9 screen we identify regulators of enhancer function as important modulators of the resistant cell state. We show that inhibition of Lsd1 (Kdm1a) is able to overcome stable epigenetic resistance by facilitating the binding of the pioneer factor, Pu.1 and cofactor, Irf8, to nucleate new enhancers that regulate the expression of key survival genes. This enhancer switching results in the re-distribution of transcriptional co-activators, including Brd4, and provides the opportunity to disable their activity and overcome epigenetic resistance. Together these findings highlight key principles to help counteract non-genetic drug resistance.
Conflict of interest statement
B.E.K., O.B., H.P.M., R.G.K., A.D., N.K., and R.K.P. are employees of GlaxoSmithKline. M.A.D. has been a member of advisory boards for CTX CRC, Storm Therapeutics, Celgene and Cambridge Epigenetix. The remaining authors declare no competing interests.
Figures
Similar articles
-
RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia.Nat Commun. 2018 Mar 21;9(1):1163. doi: 10.1038/s41467-018-03513-4. Nat Commun. 2018. PMID: 29563491 Free PMC article.
-
Transcriptional plasticity promotes primary and acquired resistance to BET inhibition.Nature. 2015 Sep 24;525(7570):543-547. doi: 10.1038/nature14898. Epub 2015 Sep 14. Nature. 2015. PMID: 26367798 Free PMC article.
-
Epigenetic regulation of noncanonical menin targets modulates menin inhibitor response in acute myeloid leukemia.Blood. 2024 Nov 7;144(19):2018-2032. doi: 10.1182/blood.2023023644. Blood. 2024. PMID: 39158067 Free PMC article.
-
Perspectives for therapeutic targeting of gene mutations in acute myeloid leukaemia with normal cytogenetics.Br J Haematol. 2015 Aug;170(3):305-22. doi: 10.1111/bjh.13409. Epub 2015 Apr 19. Br J Haematol. 2015. PMID: 25891481 Review.
-
Dissecting the role of the CXCL12/CXCR4 axis in acute myeloid leukaemia.Br J Haematol. 2020 Jun;189(5):815-825. doi: 10.1111/bjh.16456. Epub 2020 Mar 5. Br J Haematol. 2020. PMID: 32135579 Review.
Cited by
-
Non-genetic mechanisms of therapeutic resistance in cancer.Nat Rev Cancer. 2020 Dec;20(12):743-756. doi: 10.1038/s41568-020-00302-4. Epub 2020 Oct 8. Nat Rev Cancer. 2020. PMID: 33033407 Review.
-
A heritable, non-genetic road to cancer evolution.Nature. 2022 Jan;601(7891):31-32. doi: 10.1038/d41586-021-03606-z. Nature. 2022. PMID: 34880486 No abstract available.
-
Gene Transcription as a Therapeutic Target in Leukemia.Int J Mol Sci. 2021 Jul 8;22(14):7340. doi: 10.3390/ijms22147340. Int J Mol Sci. 2021. PMID: 34298959 Free PMC article. Review.
-
Targeting Chromatin Complexes in Myeloid Malignancies and Beyond: From Basic Mechanisms to Clinical Innovation.Cells. 2020 Dec 21;9(12):2721. doi: 10.3390/cells9122721. Cells. 2020. PMID: 33371192 Free PMC article. Review.
-
Interplay and cooperation between GLI2 and master transcription factors promote progression of esophageal squamous cell carcinoma.Am J Hum Genet. 2025 May 1;112(5):1039-1061. doi: 10.1016/j.ajhg.2025.03.001. Epub 2025 Mar 28. Am J Hum Genet. 2025. PMID: 40157366
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
