

Establishing microbial composition measurement standards with reference frames
- PMID: 31222023
- PMCID: PMC6586903
- DOI: 10.1038/s41467-019-10656-5
Establishing microbial composition measurement standards with reference frames
Abstract
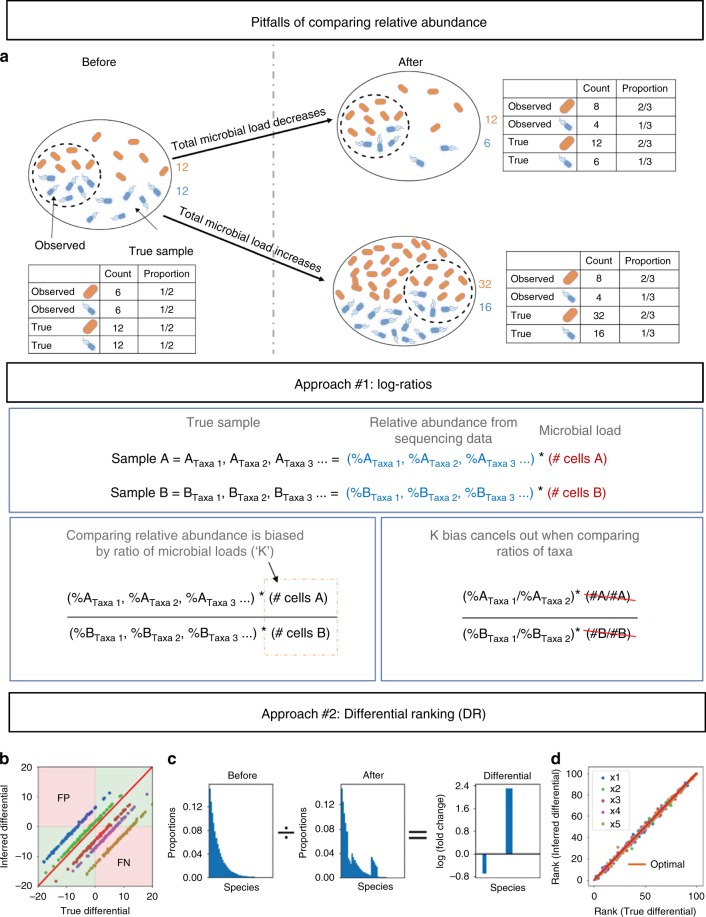
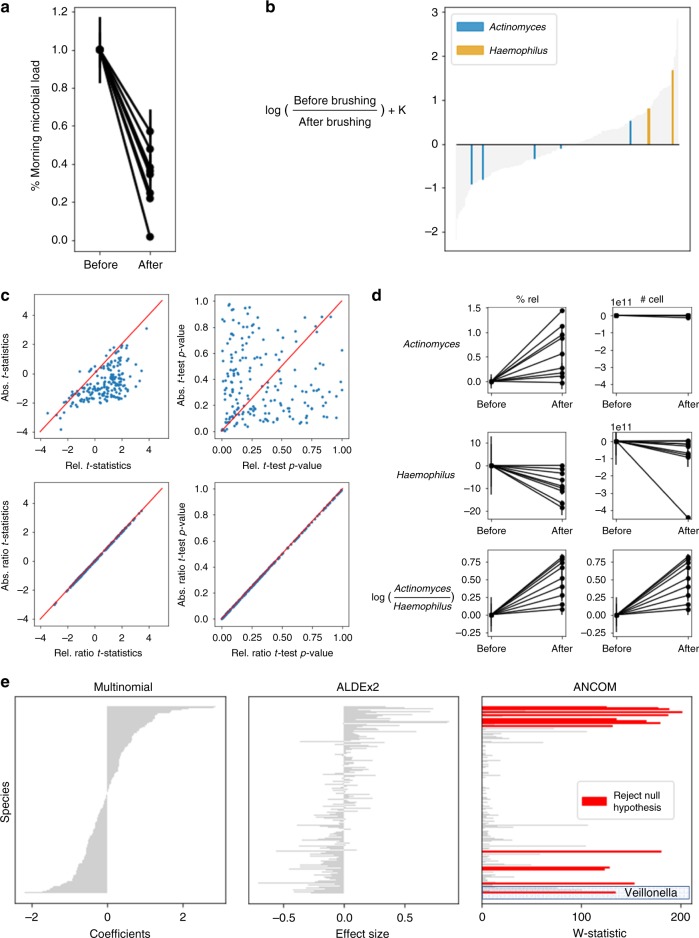
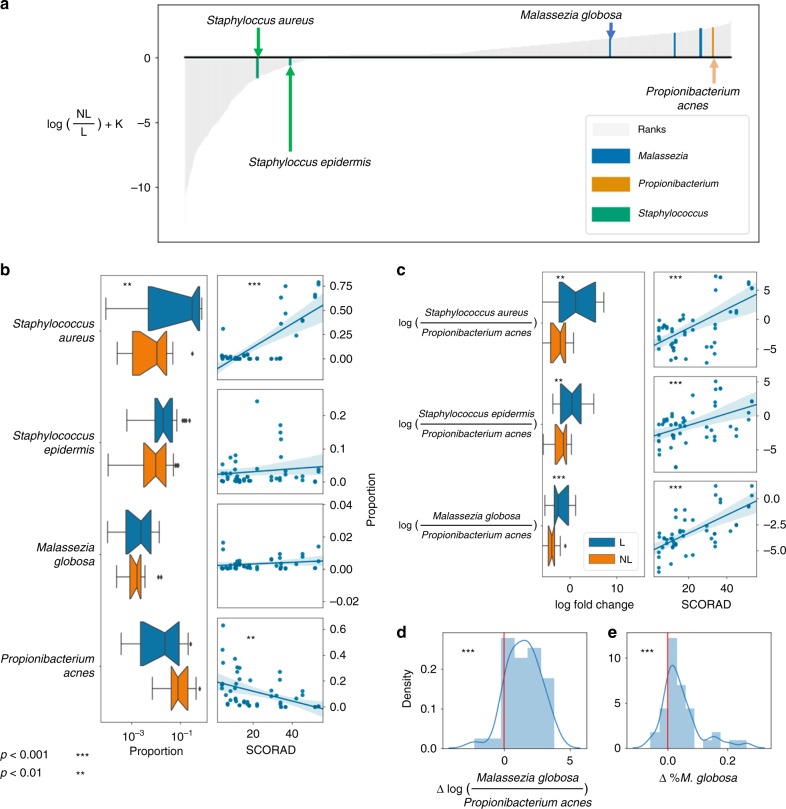
Differential abundance analysis is controversial throughout microbiome research. Gold standard approaches require laborious measurements of total microbial load, or absolute number of microorganisms, to accurately determine taxonomic shifts. Therefore, most studies rely on relative abundance data. Here, we demonstrate common pitfalls in comparing relative abundance across samples and identify two solutions that reveal microbial changes without the need to estimate total microbial load. We define the notion of "reference frames", which provide deep intuition about the compositional nature of microbiome data. In an oral time series experiment, reference frames alleviate false positives and produce consistent results on both raw and cell-count normalized data. Furthermore, reference frames identify consistent, differentially abundant microbes previously undetected in two independent published datasets from subjects with atopic dermatitis. These methods allow reassessment of published relative abundance data to reveal reproducible microbial changes from standard sequencing output without the need for new assays.
Conflict of interest statement
The authors declare no competing interests.
Figures

Comment in
-
More than microbial relative abundances.Nat Methods. 2019 Aug;16(8):678. doi: 10.1038/s41592-019-0527-3. Nat Methods. 2019. PMID: 31363209 No abstract available.
References
-
- Russel, J. et al. Datest: a framework for choosing differential abundance or expression method. Preprint at bioRxiv 10.1101/241802v 1241802 (2018).
-
- Hawinkel, S., Mattiello, F., Bijnens, L. & Thas, O. A broken promise: microbiome differential abundance methods do not control the false discovery rate. Brief. Bioinform. 20, 210–221 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- GRFP DGE-1144086/National Science Foundation (NSF)/International
- 1F31DE028478/U.S. Department of Health & Human Services | NIH | National Institute of Dental and Craniofacial Research (NIDCR)/International
- 20175015/Janssen Pharmaceuticals (Janssen Pharmaceuticals, Inc.)/International
- G-2017-9838/Alfred P. Sloan Foundation/International
- DBI-1565057/National Science Foundation (NSF)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
