

High-Resolution Copy Number Patterns From Clinically Relevant FFPE Material
- PMID: 31222134
- PMCID: PMC6586881
- DOI: 10.1038/s41598-019-45210-2
High-Resolution Copy Number Patterns From Clinically Relevant FFPE Material
Abstract
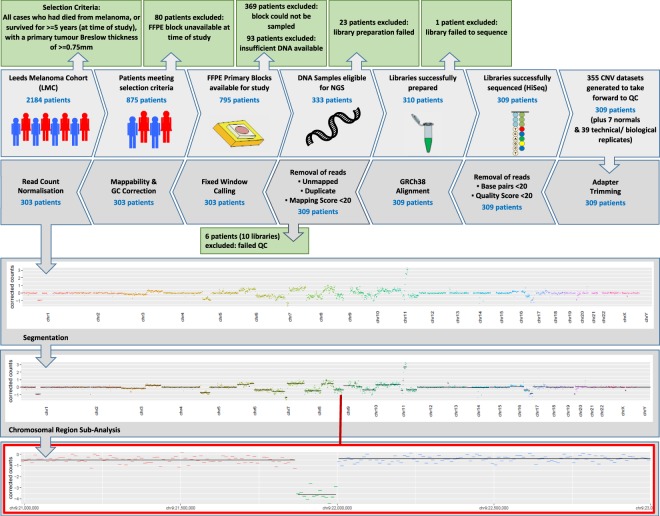
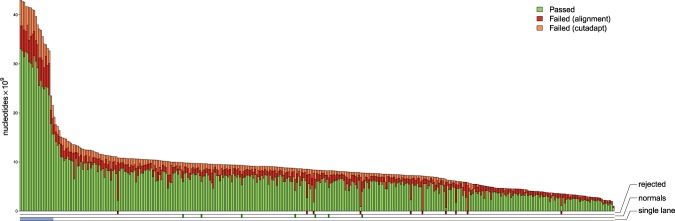
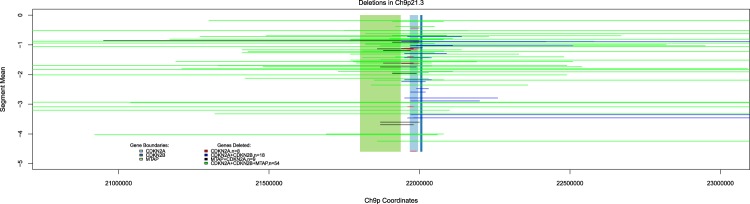
Systematic tumour profiling is essential for biomarker research and clinically for assessing response to therapy. Solving the challenge of delivering informative copy number (CN) profiles from formalin-fixed paraffin embedded (FFPE) material, the only likely readily available biospecimen for most cancers, involves successful processing of small quantities of degraded DNA. To investigate the potential for analysis of such lesions, whole-genome CNVseq was applied to 300 FFPE primary tumour samples, obtained from a large-scale epidemiological study of melanoma. The quality and the discriminatory power of CNVseq was assessed. Libraries were successfully generated for 93% of blocks, with input DNA quantity being the only predictor of success (success rate dropped to 65% if <20 ng available); 3% of libraries were dropped because of low sequence alignment rates. Technical replicates showed high reproducibility. Comparison with targeted CN assessment showed consistency with the Next Generation Sequencing (NGS) analysis. We were able to detect and distinguish CN changes with a resolution of ≤10 kb. To demonstrate performance, we report the spectrum of genomic CN alterations (CNAs) detected at 9p21, the major site of CN change in melanoma. This successful analysis of CN in FFPE material using NGS provides proof of principle for intensive examination of population-based samples.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
