

Metabolic Intermediates in Tumorigenesis and Progression
- PMID: 31223279
- PMCID: PMC6567815
- DOI: 10.7150/ijbs.33496
Metabolic Intermediates in Tumorigenesis and Progression
Abstract
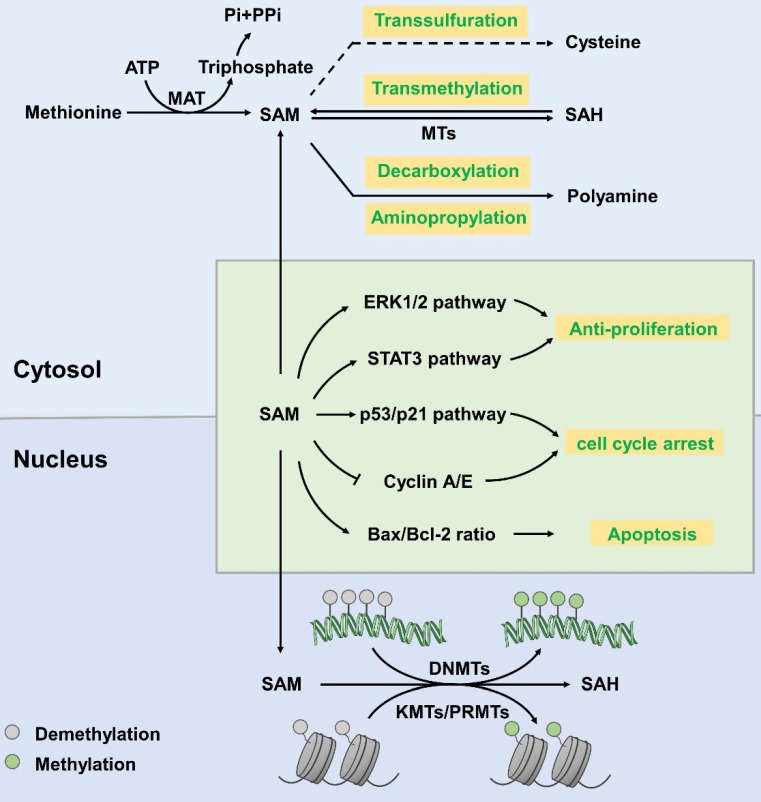
Traditional antitumor drugs inhibit the proliferation and metastasis of tumour cells by restraining the replication and expression of DNA. These drugs are usually highly cytotoxic. They kill tumour cells while also cause damage to normal cells at the same time, especially the hematopoietic cells that divide vigorously. Patients are exposed to other serious situations such as a severe infection caused by a decrease in the number of white blood cells. Energy metabolism is an essential process for the survival of all cells, but differs greatly between normal cells and tumour cells in metabolic pathways and metabolic intermediates. Whether this difference could be used as new therapeutic target while reducing damage to normal tissues is the topic of this paper. In this paper, we introduce five major metabolic intermediates in detail, including acetyl-CoA, SAM, FAD, NAD+ and THF. Their contents and functions in tumour cells and normal cells are significantly different. And the possible regulatory mechanisms that lead to these differences are proposed carefully. It is hoped that the key enzymes in these regulatory pathways could be used as new targets for tumour therapy.
Keywords: FAD; NAD+; SAM; THF; acetyl-CoA; cancer.
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures




Similar articles
-
Efficient incorporation of CoA, NAD and FAD into RNA by in vitro transcription.Nucleic Acids Res. 2003 Feb 1;31(3):e8. doi: 10.1093/nar/gng008. Nucleic Acids Res. 2003. PMID: 12560511 Free PMC article.
-
Adenine recognition: a motif present in ATP-, CoA-, NAD-, NADP-, and FAD-dependent proteins.Proteins. 2001 Aug 15;44(3):282-91. doi: 10.1002/prot.1093. Proteins. 2001. PMID: 11455601
-
Rossmann-Fold Methyltransferases: Taking a "β-Turn" around Their Cofactor, S-Adenosylmethionine.Biochemistry. 2019 Jan 22;58(3):166-170. doi: 10.1021/acs.biochem.8b00994. Epub 2018 Nov 15. Biochemistry. 2019. PMID: 30406995
-
Metabolism and epigenetics: a link cancer cells exploit.Curr Opin Biotechnol. 2015 Aug;34:23-9. doi: 10.1016/j.copbio.2014.11.012. Epub 2014 Nov 29. Curr Opin Biotechnol. 2015. PMID: 25461508 Free PMC article. Review.
-
Flavin-Dependent Methylation of RNAs: Complex Chemistry for a Simple Modification.J Mol Biol. 2016 Dec 4;428(24 Pt B):4867-4881. doi: 10.1016/j.jmb.2016.10.031. Epub 2016 Nov 5. J Mol Biol. 2016. PMID: 27825927 Review.
Cited by
-
Unveiling the Therapeutic Potential of Folate-Dependent One-Carbon Metabolism in Cancer and Neurodegeneration.Int J Mol Sci. 2024 Aug 28;25(17):9339. doi: 10.3390/ijms25179339. Int J Mol Sci. 2024. PMID: 39273288 Free PMC article. Review.
-
Correlation of several forms of folic acid with endometrial cancer: cross-sectional data from the National Health and Nutrition Examination Surveys (NHANES) 2011-2018.J Cancer Res Clin Oncol. 2023 Nov;149(15):13619-13629. doi: 10.1007/s00432-023-05177-0. Epub 2023 Jul 29. J Cancer Res Clin Oncol. 2023. PMID: 37515615 Free PMC article.
-
Amino Acid Transporters as Potential Therapeutic Targets in Thyroid Cancer.Endocrinol Metab (Seoul). 2020 Jun;35(2):227-236. doi: 10.3803/EnM.2020.35.2.227. Epub 2020 Jun 24. Endocrinol Metab (Seoul). 2020. PMID: 32615707 Free PMC article.
-
A novel seven-gene signature as Prognostic Biomarker in Hepatocellular Carcinoma.J Cancer. 2020 Jul 31;11(19):5768-5781. doi: 10.7150/jca.44573. eCollection 2020. J Cancer. 2020. PMID: 32913470 Free PMC article.
-
Nanomaterial-assisted oncolytic bacteria in solid tumor diagnosis and therapeutics.Bioeng Transl Med. 2024 Apr 17;9(4):e10672. doi: 10.1002/btm2.10672. eCollection 2024 Jul. Bioeng Transl Med. 2024. PMID: 39036084 Free PMC article. Review.
References
-
- Campbell SL, Wellen KE. Metabolic Signaling to the Nucleus in Cancer. Molecular cell. 2018;71:398–408. - PubMed
-
- Sebastian C. Tracking down the origin of cancer: metabolic reprogramming as a driver of stemness and tumorigenesis. Critical reviews in oncogenesis. 2014;19:363–82. - PubMed
-
- Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nature reviews Clinical oncology. 2017;14:11–31. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
