

Atlas of Subcellular RNA Localization Revealed by APEX-Seq
- PMID: 31230715
- PMCID: PMC6786773
- DOI: 10.1016/j.cell.2019.05.027
Atlas of Subcellular RNA Localization Revealed by APEX-Seq
Abstract
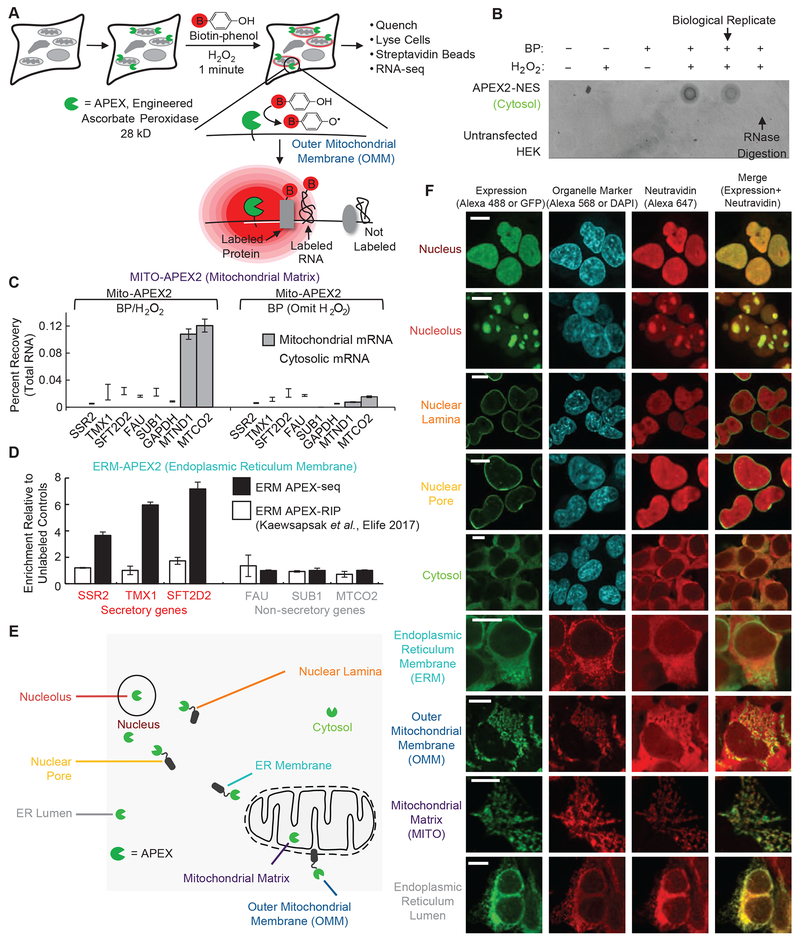
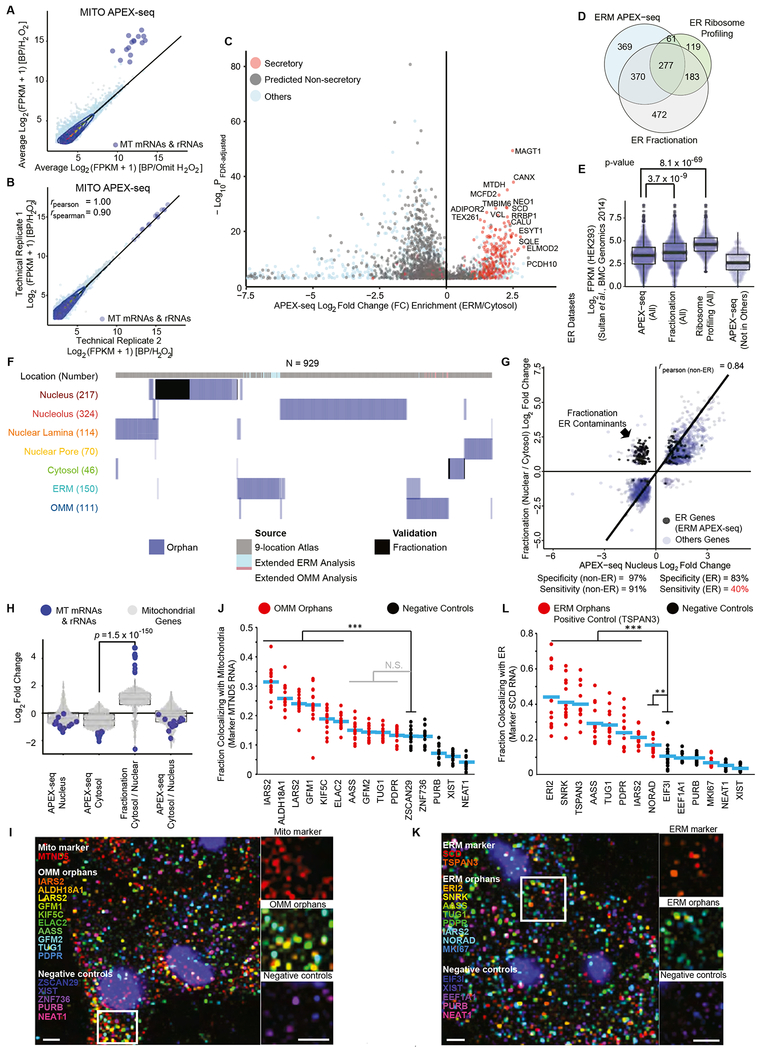
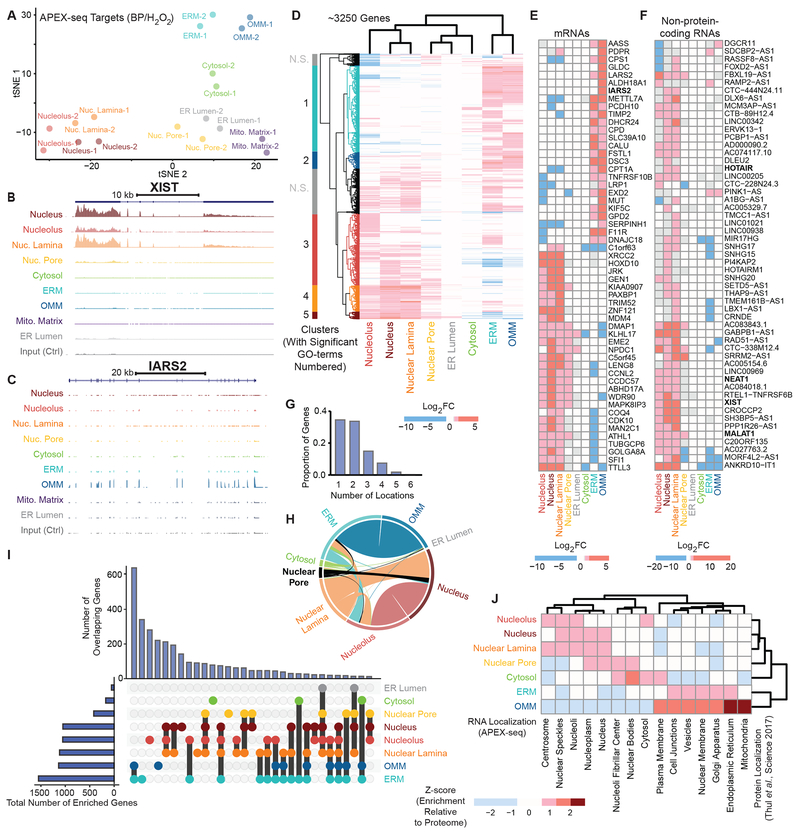
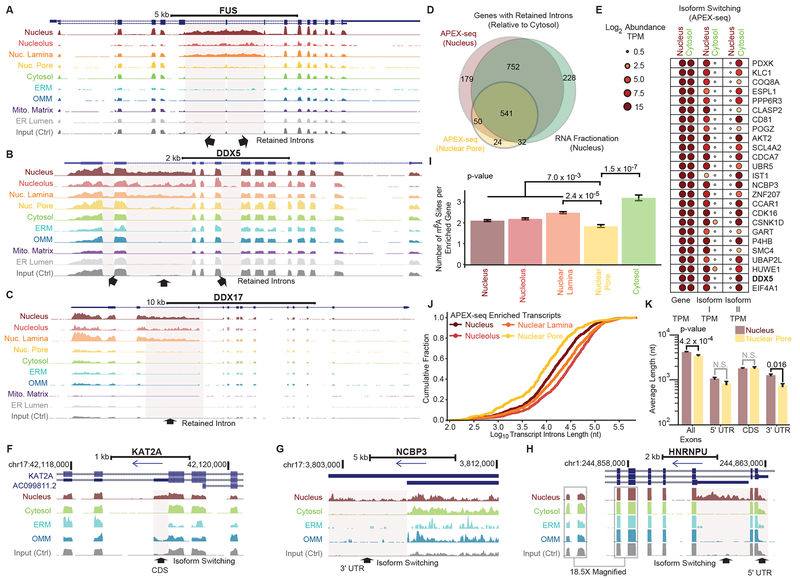
We introduce APEX-seq, a method for RNA sequencing based on direct proximity labeling of RNA using the peroxidase enzyme APEX2. APEX-seq in nine distinct subcellular locales produced a nanometer-resolution spatial map of the human transcriptome as a resource, revealing extensive patterns of localization for diverse RNA classes and transcript isoforms. We uncover a radial organization of the nuclear transcriptome, which is gated at the inner surface of the nuclear pore for cytoplasmic export of processed transcripts. We identify two distinct pathways of messenger RNA localization to mitochondria, each associated with specific sets of transcripts for building complementary macromolecular machines within the organelle. APEX-seq should be widely applicable to many systems, enabling comprehensive investigations of the spatial transcriptome.
Keywords: LADs; OXPHOS; UTRs; cycloheximide; motifs; nocodazole; retrotransposons; spatial transcriptomics; translation.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
A.Y.T., P.K., H.Y.C. and F.M.F. have filed a patent application covering aspects of this work (Patent Application Number US 2017/0226561). H.Y.C. is a co-founder and advisor of Accent Therapeutics. H.Y.C. is an advisor of 10X Genomics and Spring Discovery.
Figures


References
-
- Battich N, Stoeger T, and Pelkmans L (2015). Control of transcript variability in single mammalian cells. Cell 163, 1596–1610. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
