

The Genomics of Arthrogryposis, a Complex Trait: Candidate Genes and Further Evidence for Oligogenic Inheritance
- PMID: 31230720
- PMCID: PMC6612529
- DOI: 10.1016/j.ajhg.2019.05.015
The Genomics of Arthrogryposis, a Complex Trait: Candidate Genes and Further Evidence for Oligogenic Inheritance
Abstract
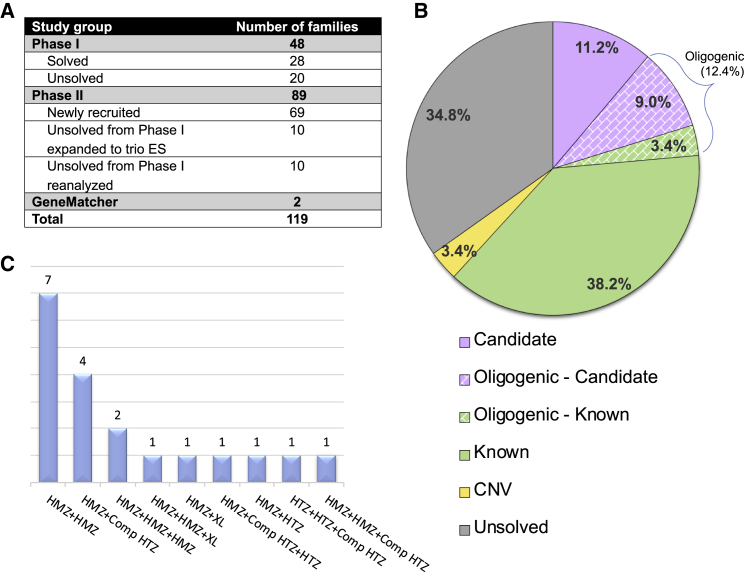
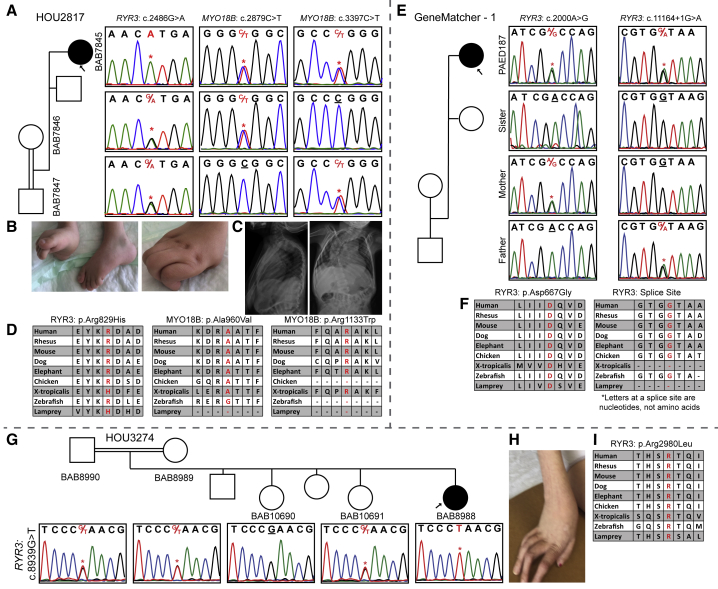
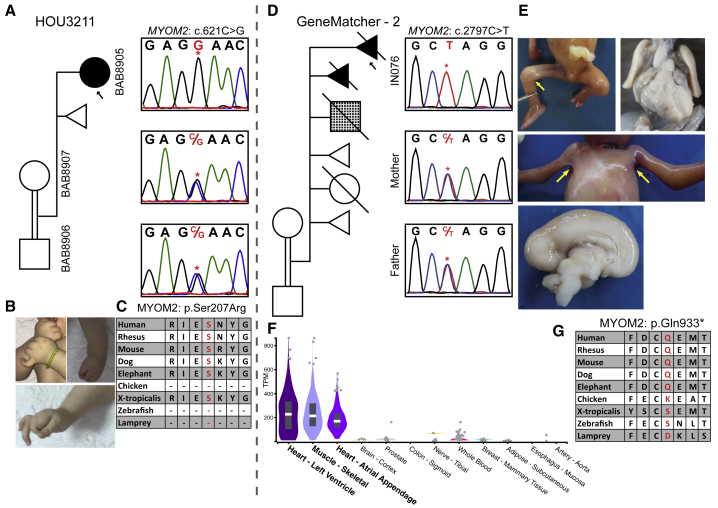
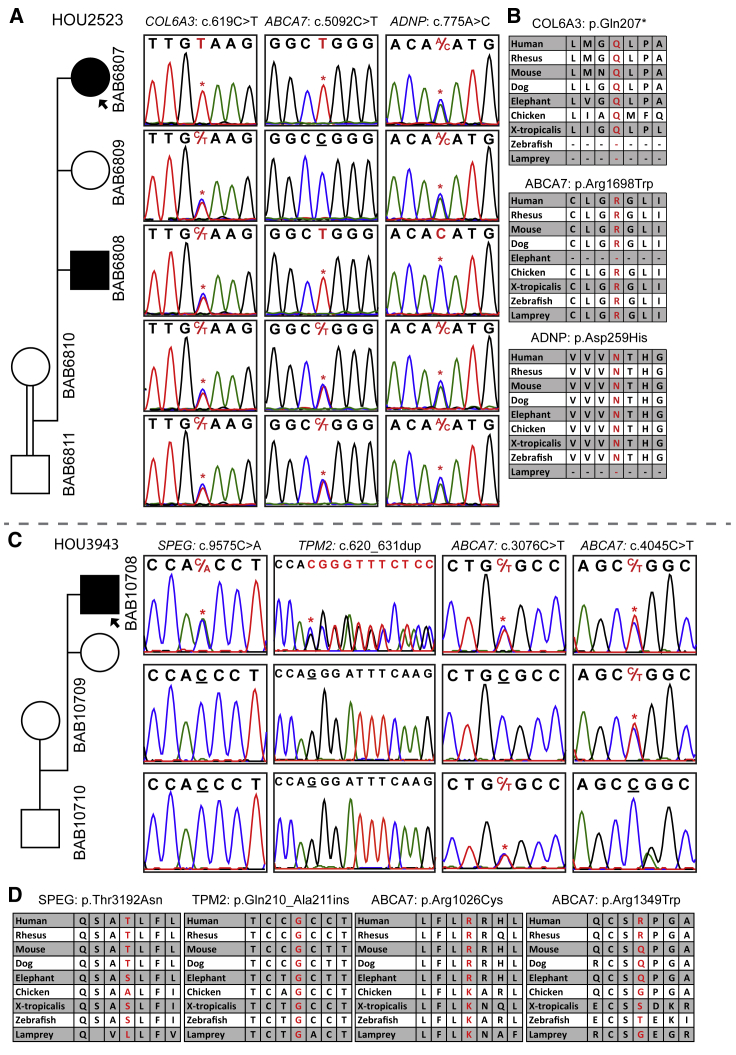
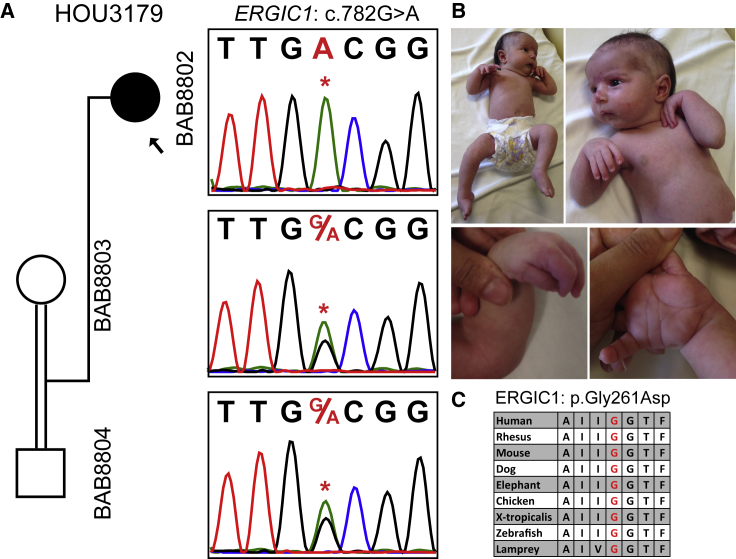
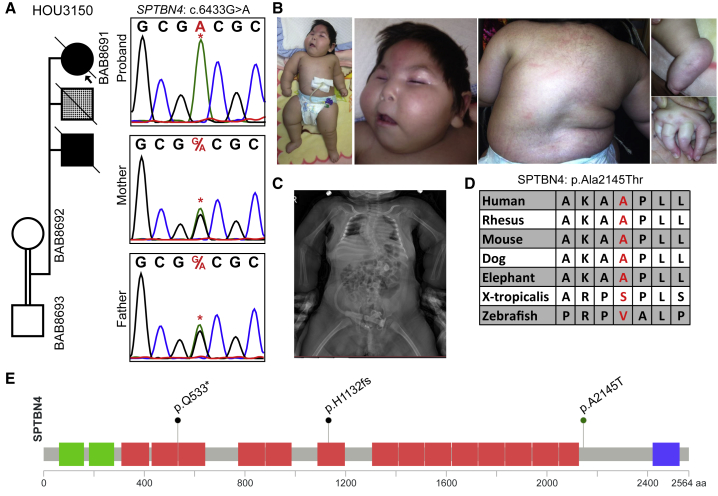
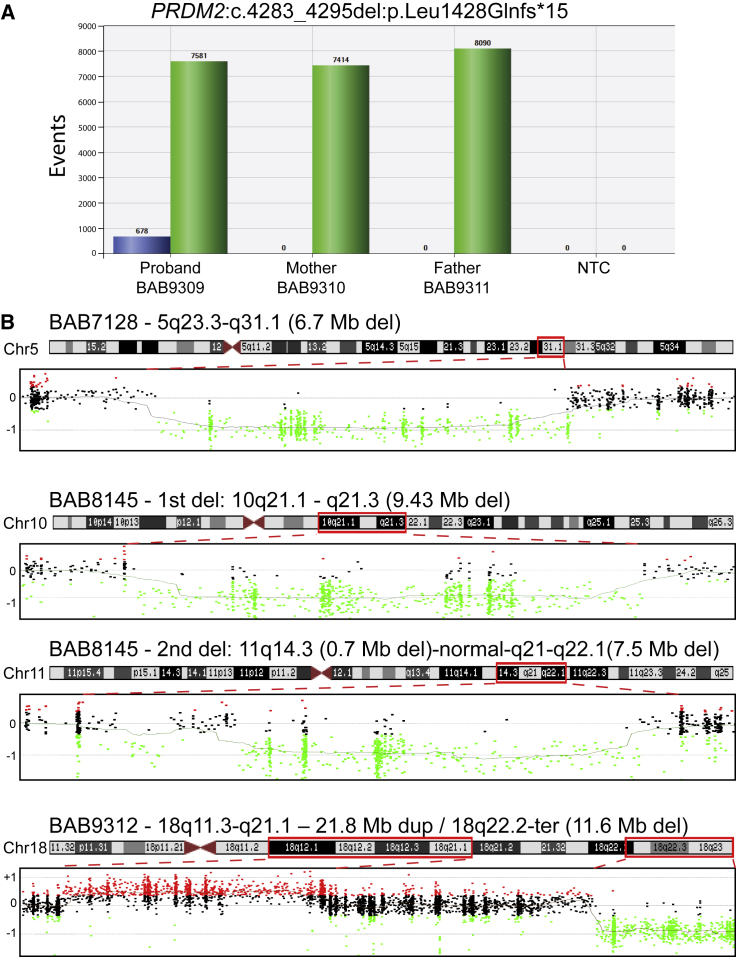
Arthrogryposis is a clinical finding that is present either as a feature of a neuromuscular condition or as part of a systemic disease in over 400 Mendelian conditions. The underlying molecular etiology remains largely unknown because of genetic and phenotypic heterogeneity. We applied exome sequencing (ES) in a cohort of 89 families with the clinical sign of arthrogryposis. Additional molecular techniques including array comparative genomic hybridization (aCGH) and Droplet Digital PCR (ddPCR) were performed on individuals who were found to have pathogenic copy number variants (CNVs) and mosaicism, respectively. A molecular diagnosis was established in 65.2% (58/89) of families. Eleven out of 58 families (19.0%) showed evidence for potential involvement of pathogenic variation at more than one locus, probably driven by absence of heterozygosity (AOH) burden due to identity-by-descent (IBD). RYR3, MYOM2, ERGIC1, SPTBN4, and ABCA7 represent genes, identified in two or more families, for which mutations are probably causative for arthrogryposis. We also provide evidence for the involvement of CNVs in the etiology of arthrogryposis and for the idea that both mono-allelic and bi-allelic variants in the same gene cause either similar or distinct syndromes. We were able to identify the molecular etiology in nine out of 20 families who underwent reanalysis. In summary, our data from family-based ES further delineate the molecular etiology of arthrogryposis, yielded several candidate disease-associated genes, and provide evidence for mutational burden in a biological pathway or network. Our study also highlights the importance of reanalysis of individuals with unsolved diagnoses in conjunction with sequencing extended family members.
Keywords: ES reanalysis; absence of heterozygosity; arthrogryposis; identity-by-descent; joint contracture; multilocus pathogenic variation; neuromuscular disease; trio-exome.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Hall J.G. Arthrogryposis (multiple congenital contractures): Diagnostic approach to etiology, classification, genetics, and general principles. Eur. J. Med. Genet. 2014;57:464–472. - PubMed
- Hall, J.G. (2014). Arthrogryposis (multiple congenital contractures): Diagnostic approach to etiology, classification, genetics, and general principles. Eur. J. Med. Genet. 57, 464-472. - PubMed
-
- Lowry R.B., Sibbald B., Bedard T., Hall J.G. Prevalence of multiple congenital contractures including arthrogryposis multiplex congenita in Alberta, Canada, and a strategy for classification and coding. Birth Defects Res. A Clin. Mol. Teratol. 2010;88:1057–1061. - PubMed
- Lowry, R.B., Sibbald, B., Bedard, T., and Hall, J.G. (2010). Prevalence of multiple congenital contractures including arthrogryposis multiplex congenita in Alberta, Canada, and a strategy for classification and coding. Birth Defects Res. A Clin. Mol. Teratol. 88, 1057-1061. - PubMed
-
- Balta B., Erdogan M., Ergul A.B., Sahin Y., Ozcan A. Interstitial deletion 5p14.1-p15.2 and 5q14.3-q23.2 in a patient with clubfoot, blepharophimosis, arthrogryposis, and multiple congenital abnormalities. Am. J. Med. Genet. A. 2017;173:2798–2802. - PubMed
- Balta, B., Erdogan, M., Ergul, A.B., Sahin, Y., and Ozcan, A. (2017). Interstitial deletion 5p14.1-p15.2 and 5q14.3-q23.2 in a patient with clubfoot, blepharophimosis, arthrogryposis, and multiple congenital abnormalities. Am. J. Med. Genet. A. 173, 2798-2802. - PubMed
-
- Carrascosa-Romero M.C., Suela J., Pardal-Fernández J.M., Bermejo-Sánchez E., Vidal-Company A., MacDonald A., Tébar-Gil R., Martínez-Fernández M.L., Martínez-Frías M.L. A 2.84 Mb deletion at 21q22.11 in a patient clinically diagnosed with Marden-Walker syndrome. Am. J. Med. Genet. A. 2013;161A:2281–2290. - PubMed
- Carrascosa-Romero, M.C., Suela, J., Pardal-Fernandez, J.M., Bermejo-Sanchez, E., Vidal-Company, A., MacDonald, A., Tebar-Gil, R., Martinez-Fernandez, M.L., and Martinez-Frias, M.L. (2013). A 2.84 Mb deletion at 21q22.11 in a patient clinically diagnosed with Marden-Walker syndrome. Am. J. Med. Genet. A. 161A, 2281-2290. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
