

De-duplicating patient records from three independent data sources reveals the incidence of rare neuromuscular disorders in Germany
- PMID: 31234869
- PMCID: PMC6591958
- DOI: 10.1186/s13023-019-1125-2
De-duplicating patient records from three independent data sources reveals the incidence of rare neuromuscular disorders in Germany
Abstract
Background: Estimation of incidence in rare diseases is often challenging due to unspecific and incomplete coding and recording systems. Patient- and health care provider-driven data collections are held with different organizations behind firewalls to protect the privacy of patients. They tend to be fragmented, incomplete and their aggregation leads to further inaccuracies, as the duplicated records cannot easily be identified. We here report about a novel approach to evaluate the incidences of Duchenne muscular dystrophy (DMD) and spinal muscular atrophy (SMA) in Germany.
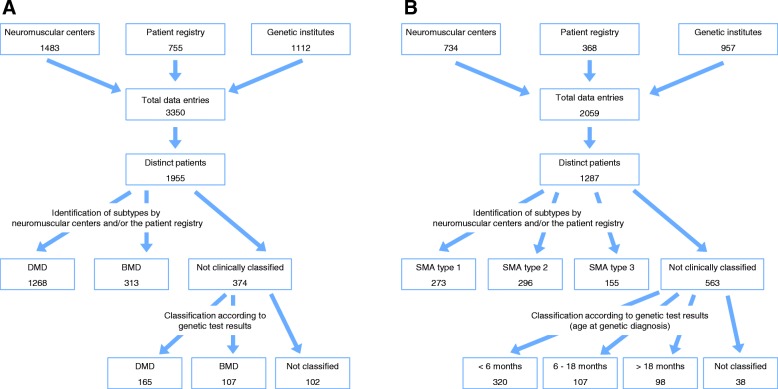
Methods: We performed a retrospective epidemiological study collecting data from patients with dystrophinopathies (DMD and Becker muscular dystrophy) and SMA born between 1995 and 2018. We invited all neuromuscular centers, genetic institutes and the patient registries for DMD and SMA in Germany to participate in the data collection. A novel web-based application for data entry was developed converting patient identifying information into a hash code. Duplicate entries were reliably allocated to the distinct patient.
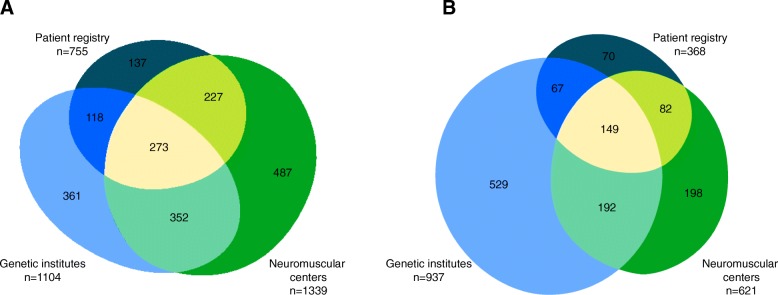
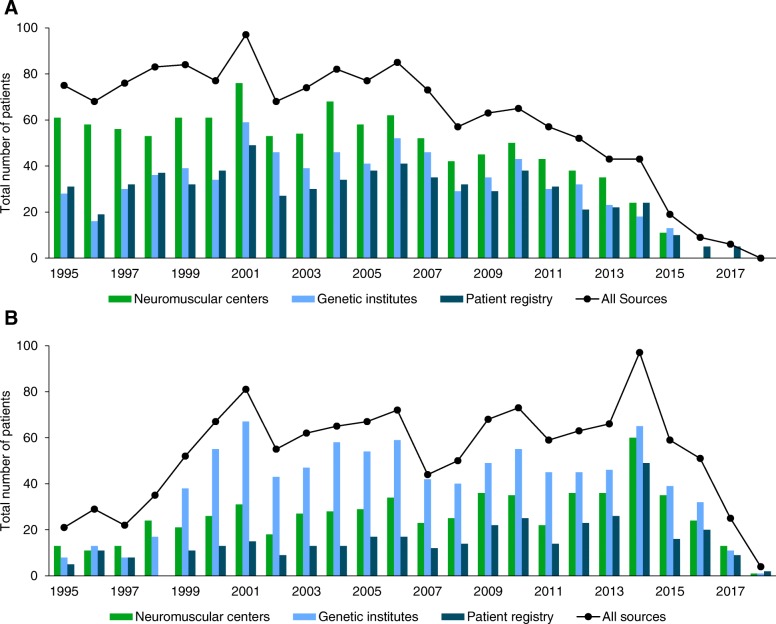
Results: We collected 5409 data entries in our web-based database representing 1955 distinct patients with dystrophinopathies and 1287 patients with SMA. 55.0% of distinct patients were found in one of the 3 data sources only, while 32.0% were found in 2, and 13.0% in all 3 data sources. The highest number of SMA patients was reported by genetic testing laboratories, while for DMD the highest number was reported by the clinical specialist centers. After the removal of duplicate records, the highest yearly incidence for DMD was calculated as 2.57:10,000 in 2001 and the highest incidence for SMA as 1.36:10,000 in 2014.
Conclusion: With our novel approach (compliant with data protection regulations), we were able to identify unique patient records and estimate the incidence of DMD and SMA in Germany combining and de-duplicating data from patient registries, genetic institutes, and clinical care centers. Although we combined three different data sources, an unknown number of patients might not have been reported by any of these sources. Therefore, our results reflect the minimal incidence of these diseases.
Keywords: Duchenne muscular dystrophy; Incidence; Neuromuscular disease; Spinal muscular atrophy.
Conflict of interest statement
JK received research funding and/or compensation for presentations and consultancy from Avexis, Biogen, Ionis Pharmaceuticals, Novartis, and Roche. HL received research funding and/or compensation for presentations and consultancy from AMO Pharma, Biogen, Desitin, Roche, Santhera, Sarepta, Satellos, Ultragenyx. AP received compensations for presentations and training activities from Biogen. DS participated in workshops sponsored by Biogen. MCW received research funding and/or compensation for presentations and consultancy from Avexis, Biogen, Grünenthal, Novartis, PTC, Roche, Santhera, and Sarepta.
Figures
References
-
- Baker D, Knoppers BM, Phillips M, van Enckevort D, Kaufmann P, Lochmuller H, Taruscio D. Privacy-preserving linkage of genomic and clinical data sets. IEEE/ACM Trans Comput Biol Bioinform. 2018. 10.1109/TCBB.2018.2855125. - PubMed
-
- Ebner H, Hayn D, Falgenhauer M, Nitzlnader M, Schleiermacher G, Haupt R, et al. Piloting the European unified patient identity management (EUPID) concept to facilitate secondary use of neuroblastoma data from Clinical Trials and Biobanking. Stud Health Technol Inform. 2016;223:31–38. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
