

Genome sequencing and implications for rare disorders
- PMID: 31234920
- PMCID: PMC6591893
- DOI: 10.1186/s13023-019-1127-0
Genome sequencing and implications for rare disorders
Abstract
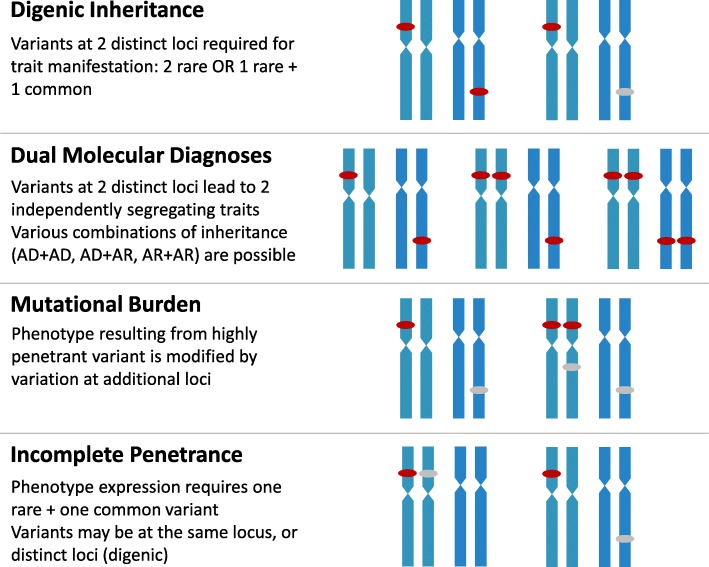
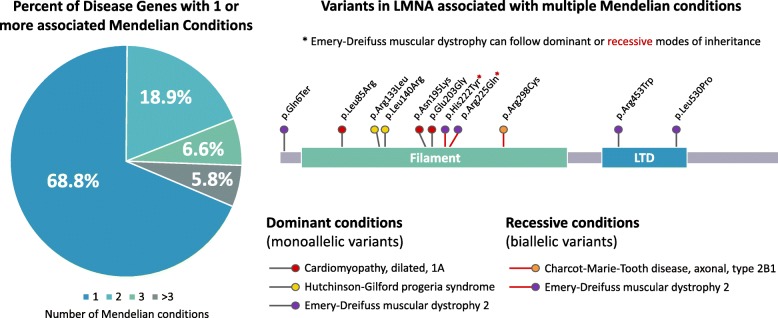
The practice of genomic medicine stands to revolutionize our approach to medical care, and to realize this goal will require discovery of the relationship between rare variation at each of the ~ 20,000 protein-coding genes and their consequent impact on individual health and expression of Mendelian disease. The step-wise evolution of broad-based, genome-wide cytogenetic and molecular genomic testing approaches (karyotyping, chromosomal microarray [CMA], exome sequencing [ES]) has driven much of the rare disease discovery to this point, with genome sequencing representing the newest member of this team. Each step has brought increased sensitivity to interrogate individual genomic variation in an unbiased method that does not require clinical prediction of the locus or loci involved. Notably, each step has also brought unique limitations in variant detection, for example, the low sensitivity of ES for detection of triploidy, and of CMA for detection of copy neutral structural variants. The utility of genome sequencing (GS) as a clinical molecular diagnostic test, and the increased sensitivity afforded by addition of long-read sequencing or other -omics technologies such as RNAseq or metabolomics, are not yet fully explored, though recent work supports improved sensitivity of variant detection, at least in a subset of cases. The utility of GS will also rely upon further elucidation of the complexities of genetic and allelic heterogeneity, multilocus rare variation, and the impact of rare and common variation at a locus, as well as advances in functional annotation of identified variants. Much discovery remains to be done before the potential utility of GS is fully appreciated.
Keywords: Diagnostic utility; Exome sequencing; Genome sequencing; Mendelian conditions; Molecular diagnoses; Rare disease; Undiagnosed diseases.
Conflict of interest statement
JEP is an employee of the Department of Molecular and Human Genetics at Baylor College of Medicine (BCM). BCM and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), which performs clinical exome sequencing and chromosomal microarray genomics assay services.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
