

PLA2G4A/cPLA2-mediated lysosomal membrane damage leads to inhibition of autophagy and neurodegeneration after brain trauma
- PMID: 31238788
- PMCID: PMC6999646
- DOI: 10.1080/15548627.2019.1628538
PLA2G4A/cPLA2-mediated lysosomal membrane damage leads to inhibition of autophagy and neurodegeneration after brain trauma
Abstract
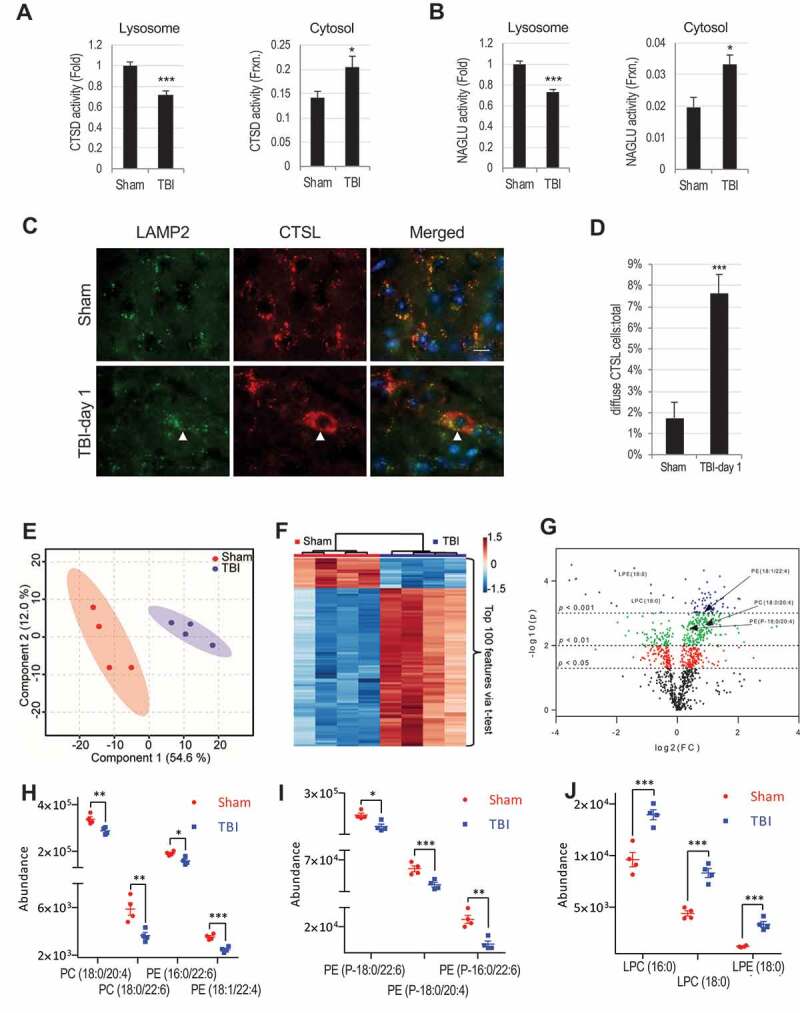
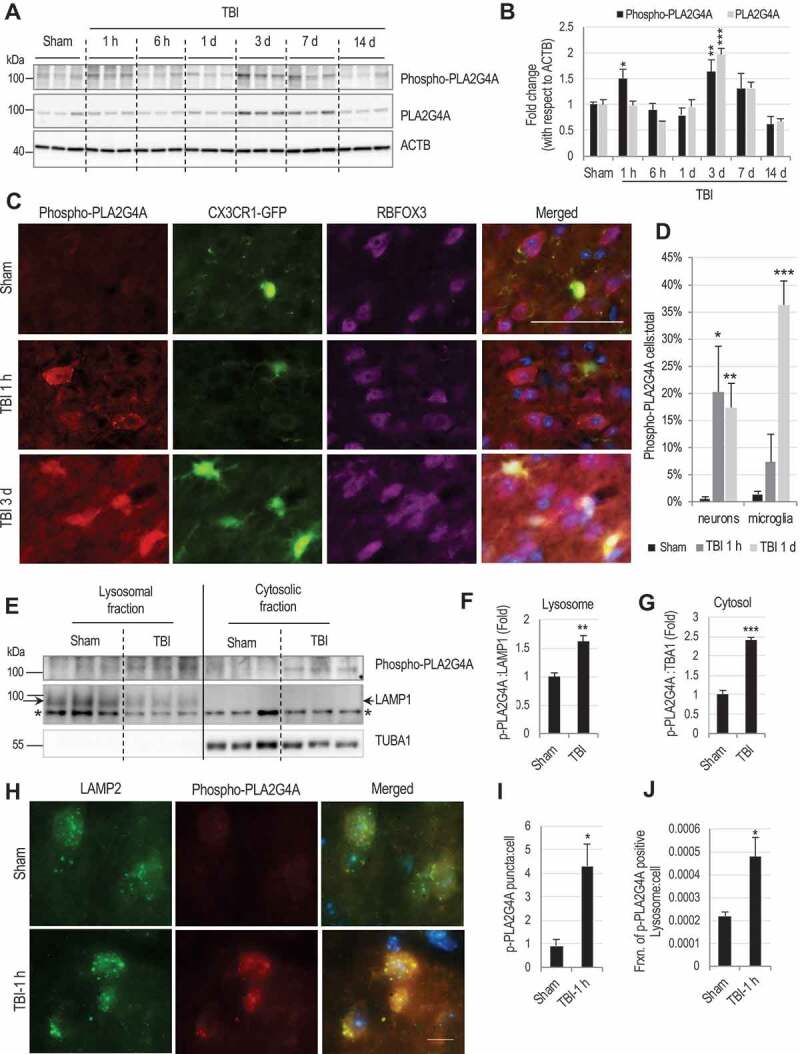
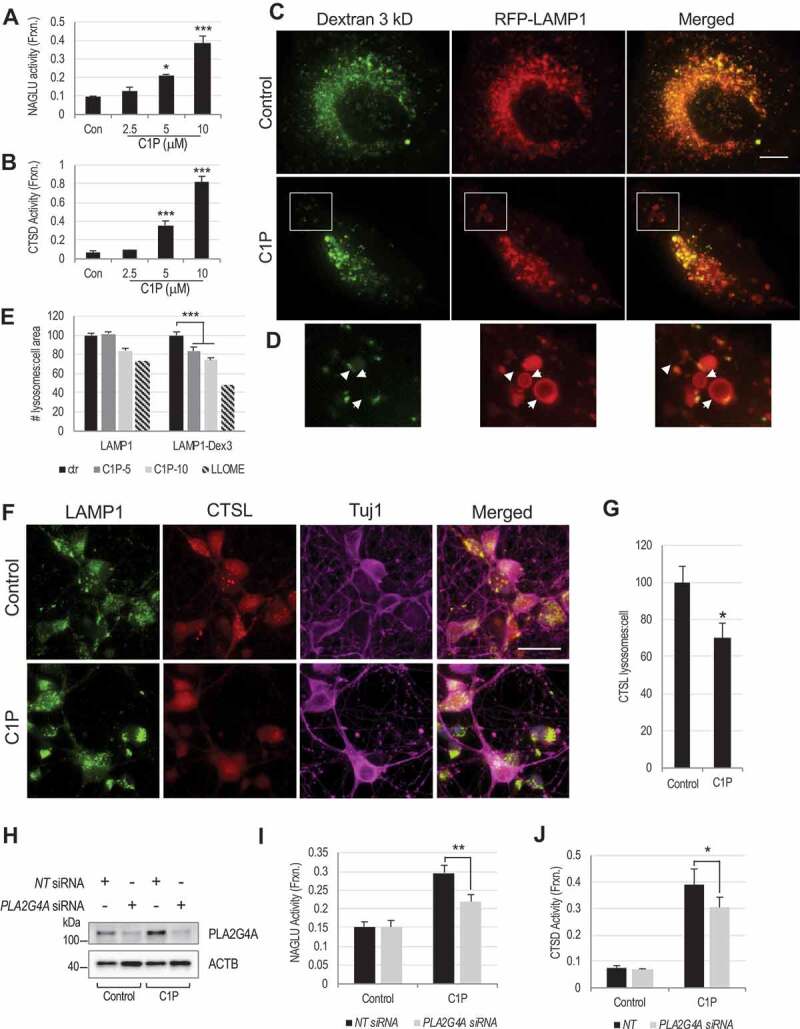
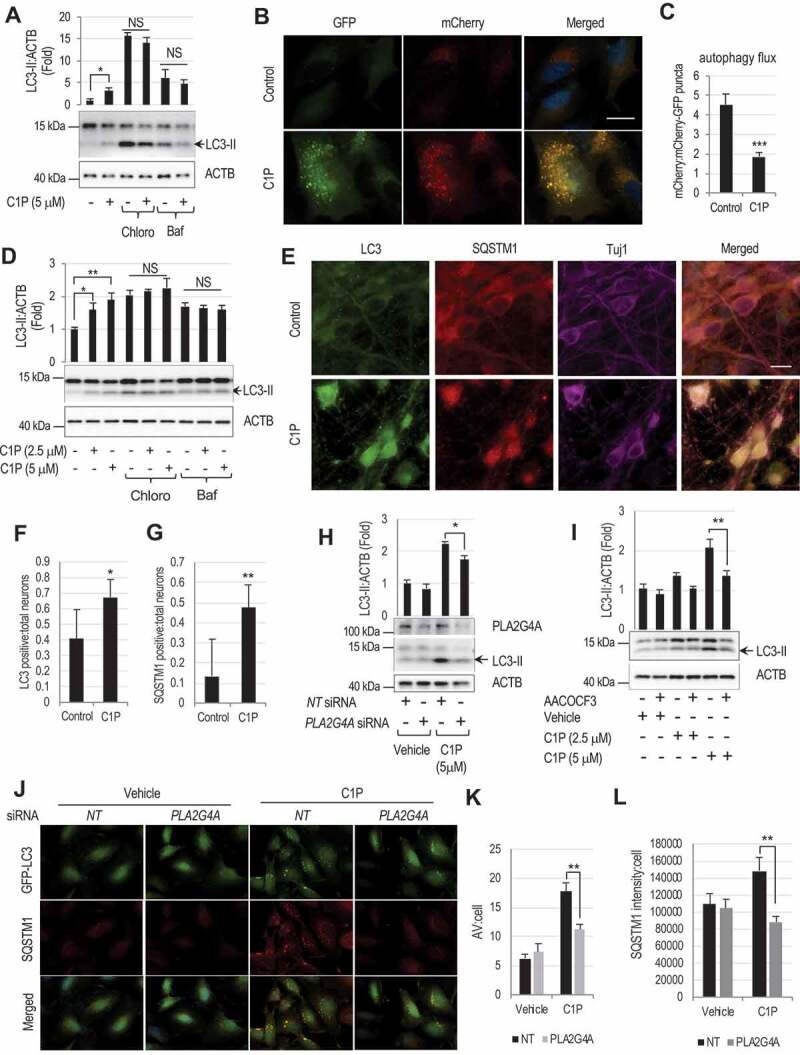
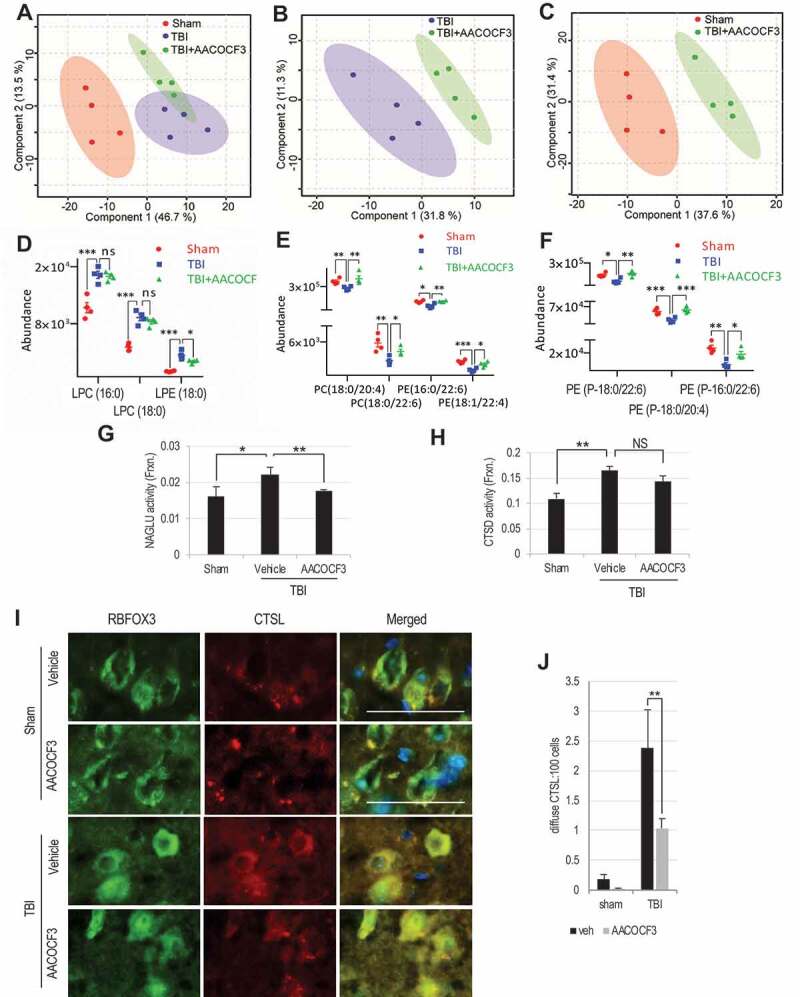
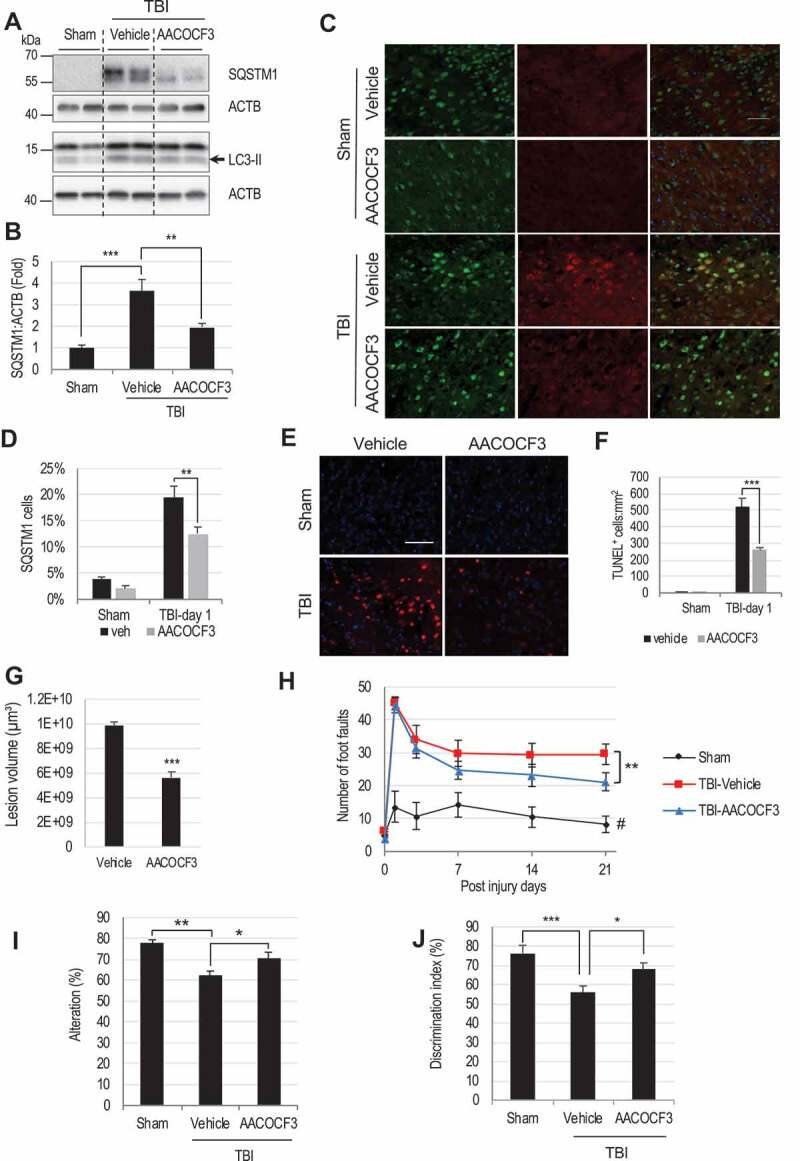
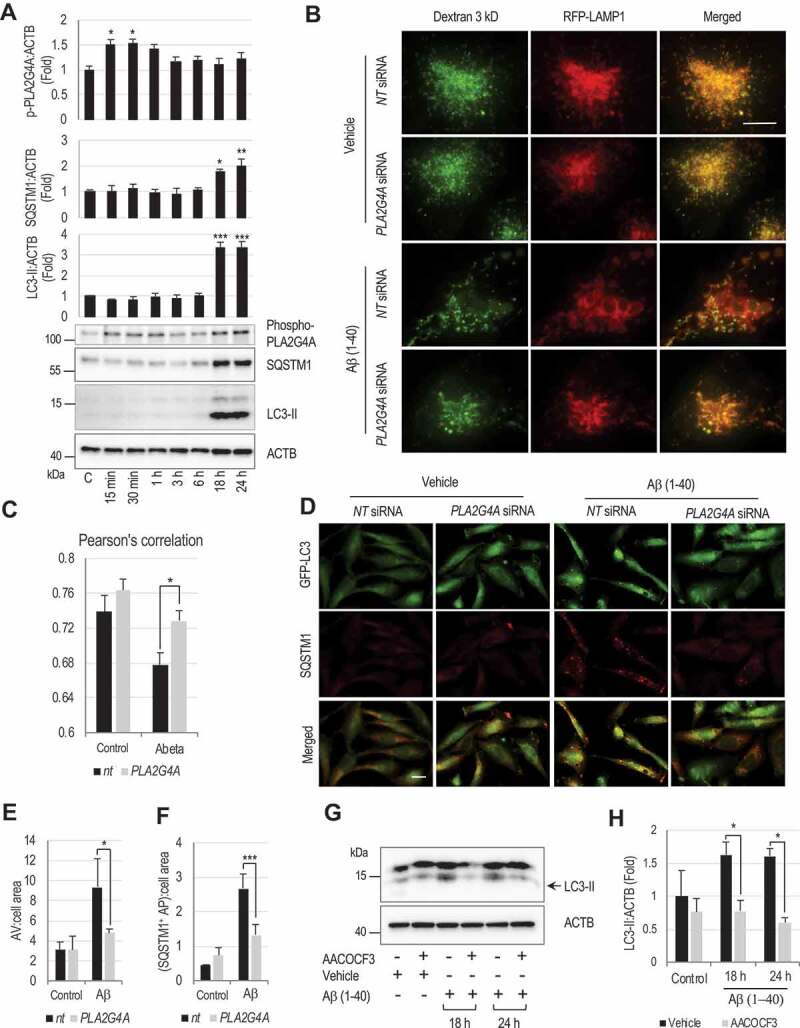
Lysosomal membrane permeabilization (LMP) is observed under many pathological conditions, leading to cellular dysfunction and death. However, the mechanisms by which lysosomal membranes become leaky in vivo are not clear. Our data demonstrate that LMP occurs in neurons following controlled cortical impact induced (CCI) traumatic brain injury (TBI) in mice, leading to impaired macroautophagy (autophagy) and neuronal cell death. Comparison of LC-MS/MS lysosomal membrane lipid profiles from TBI and sham animals suggested a role for PLA2G4A/cPLA2 (phospholipase A2, group IVA [cytosolic, calcium-dependent]) in TBI-induced LMP. Activation of PLA2G4A caused LMP and inhibition of autophagy flux in cell lines and primary neurons. In vivo pharmacological inhibition of PLA2G4A attenuated TBI-induced LMP, as well as subsequent impairment of autophagy and neuronal loss, and was associated with improved neurological outcomes. Inhibition of PLA2G4A in vitro limited amyloid-β-induced LMP and inhibition of autophagy. Together, our data indicate that PLA2G4A -mediated lysosomal membrane damage is involved in neuronal cell death following CCI-induced TBI and potentially in other neurodegenerative disorders.Abbreviations: AACOCF3, arachidonyl trifluoromethyl ketone; ACTB/β-actin, actin, beta; AD, Alzheimer disease; ATG5, autophagy related 5; ATG7, autophagy related 7; ATG12, autophagy related 12; BECN1, beclin 1, autophagy related; C1P, ceramide-1-phosphate; CCI, controlled cortical impact; CTSD, cathepsin D; CTSL, cathepsin L; GFP, green fluorescent protein; IF, immunofluorescence; LAMP1, lysosomal-associated membrane protein 1; LAMP2, lysosomal-associated membrane protein 2; LC-MS/MS, liquid chromatography-tandem mass spectrometry; LMP, Lysosomal membrane permeabilization; LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; MAP1LC3/LC3, microtuble-associated protein 1 light chain 3; NAGLU, alpha-N-acetylglucosaminidase (Sanfilippo disease IIIB); PC, diacyl glycerophosphatidylcholine; PE, diacyl glycerophosphatidylethanolamine; PE-O, plasmanyl glycerophosphatidylethanolamine; PE-P, plasmenyl glycerophosphatidylethanolamine; PLA2G4A/cPLA2, phospholipase A2, group IVA (cytosolic, calcium-dependent); RBFOX3, RNA binding protein, fox-1 homolog (C. elegans) 3; RFP, red fluorescent protein; ROS, reactive oxygen species; SQSTM1, sequestosome 1; TUBA1/α-tubulin, tubulin, alpha; TBI, traumatic brain injury; TFEB, transcription factor EB; ULK1, unc-51 like kinase 1.
Keywords: Amyloid β; autophagy; cytosolic phospholipase A2 (cPLA2); lysosomal membrane permeabilization (LMP); membrane lipidomic analysis; traumatic brain injury (TBI).
Figures
References
-
- Werner C, Engelhard K.. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007. July;99(1):4–9. 99/1/4 [pii] PubMed PMID: 17573392; eng. - PubMed
-
- Gardner AJ, Zafonte R. Neuroepidemiology of traumatic brain injury. Handb Clin Neurol. 2016;138:207–223. B978-0-12-802973-2.00012-4 [pii]. PubMed PMID: 27637960; eng. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous