

Compound phenotype of osteogenesis imperfecta and Ehlers-Danlos syndrome caused by combined mutations in COL1A1 and COL5A1
- PMID: 31239369
- PMCID: PMC6658722
- DOI: 10.1042/BSR20181409
Compound phenotype of osteogenesis imperfecta and Ehlers-Danlos syndrome caused by combined mutations in COL1A1 and COL5A1
Abstract
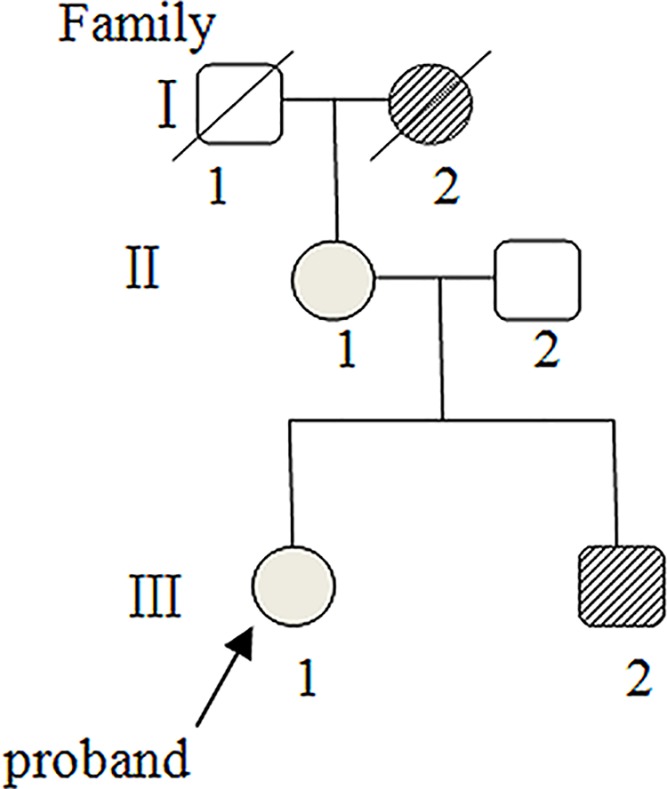
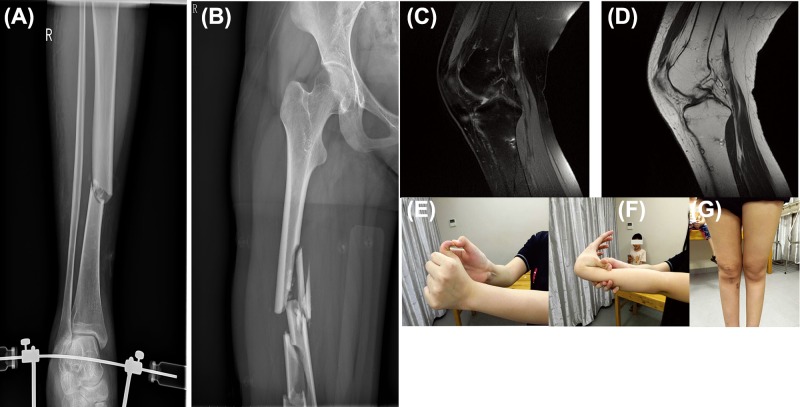
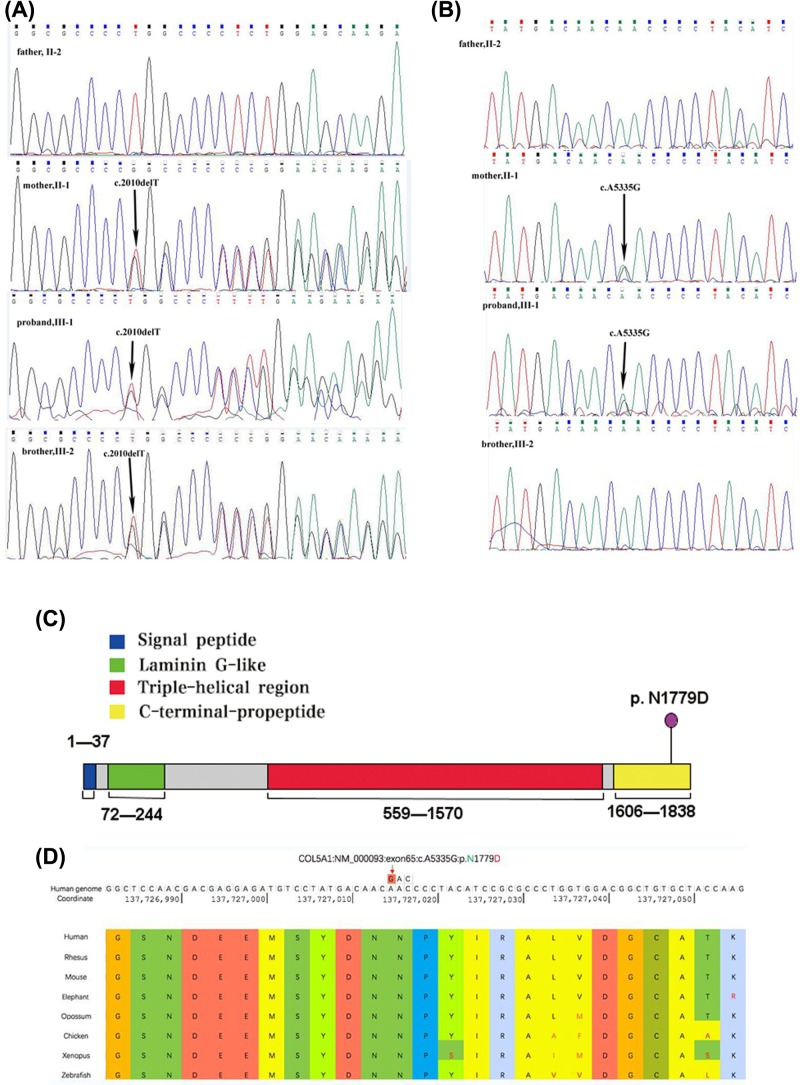
Osteogenesis imperfecta (OI) is an inherited connective tissue disorder with a broad clinical spectrum that can overlap with Ehlers-Danlos syndrome (EDS). To date, patients with both OI and EDS have rarely been reported. In the present study, we investigated a family with four members, one healthy individual, one displaying OI only, and two displaying the compound phenotype of OI and EDS, and identified the pathogenic mutations. Whole exome sequencing was applied to the proband and her brother. To verify that the mutations were responsible for the pathogenesis, conventional Sanger sequencing was performed for all members of the family. We identified a known COL1A1 (encoding collagen type I α 1 chain) mutation (c.2010delT, p.Gly671Alafs*95) in all three patients (the proband, her brother, and her mother) in this family, but also a novel heterozygous COL5A1 (encoding collagen type V α 1 chain) mutation (c.5335A>G, p.N1779D) in the region encoding the C-terminal propeptide domain in the proband and her mother, who both had the compound phenotype of OI and EDS. The results of the present study suggested that the proband and her mother presented with the compound OI-EDS phenotype caused by pathogenic mutations in COL5A1 and COL1A1.
Keywords: COL1A1; COL5A1; Ehlers-Danlos syndrome; Osteogenesis imperfecta; Whole exome sequencing.
© 2019 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures
Similar articles
-
Osteogenesis Imperfecta/Ehlers-Danlos Overlap Syndrome and Neuroblastoma-Case Report and Review of Literature.Genes (Basel). 2022 Mar 25;13(4):581. doi: 10.3390/genes13040581. Genes (Basel). 2022. PMID: 35456387 Free PMC article. Review.
-
An overlapping phenotype of Osteogenesis imperfecta and Ehlers-Danlos syndrome due to a heterozygous mutation in COL1A1 and biallelic missense variants in TNXB identified by whole exome sequencing.Am J Med Genet A. 2016 Apr;170A(4):1080-5. doi: 10.1002/ajmg.a.37547. Epub 2016 Jan 22. Am J Med Genet A. 2016. PMID: 26799614
-
Familial Ehlers-Danlos syndrome with lethal arterial events caused by a mutation in COL5A1.Am J Med Genet A. 2015 Jun;167(6):1196-203. doi: 10.1002/ajmg.a.36997. Epub 2015 Apr 2. Am J Med Genet A. 2015. PMID: 25845371
-
COL1-related overlap disorder: A novel connective tissue disorder incorporating the osteogenesis imperfecta/Ehlers-Danlos syndrome overlap.Clin Genet. 2020 Mar;97(3):396-406. doi: 10.1111/cge.13683. Epub 2019 Dec 12. Clin Genet. 2020. PMID: 31794058
-
Delineation of Ehlers-Danlos syndrome phenotype due to the c.934C>T, p.(Arg312Cys) mutation in COL1A1: Report on a three-generation family without cardiovascular events, and literature review.Am J Med Genet A. 2017 Feb;173(2):524-530. doi: 10.1002/ajmg.a.38035. Epub 2016 Nov 7. Am J Med Genet A. 2017. PMID: 28102596 Review.
Cited by
-
COL1-Related Disorders: Case Report and Review of Overlapping Syndromes.Front Genet. 2021 May 7;12:640558. doi: 10.3389/fgene.2021.640558. eCollection 2021. Front Genet. 2021. PMID: 34025714 Free PMC article.
-
Integrative analyses of genetic characteristics associated with skeletal endothelial cells.Braz J Med Biol Res. 2024 Apr 19;57:e13339. doi: 10.1590/1414-431X2024e13339. eCollection 2024. Braz J Med Biol Res. 2024. PMID: 38656074 Free PMC article.
-
Collagen type 1 alpha 1 chain is a novel predictive biomarker of poor progression-free survival and chemoresistance in metastatic lung cancer.J Cancer. 2021 Jul 25;12(19):5723-5731. doi: 10.7150/jca.59723. eCollection 2021. J Cancer. 2021. PMID: 34475986 Free PMC article.
-
COL5A1 RS12722 Is Associated with Temporomandibular Joint Anterior Disc Displacement without Reduction in Polish Caucasians.Cells. 2021 Sep 14;10(9):2423. doi: 10.3390/cells10092423. Cells. 2021. PMID: 34572072 Free PMC article.
-
Osteogenesis Imperfecta/Ehlers-Danlos Overlap Syndrome and Neuroblastoma-Case Report and Review of Literature.Genes (Basel). 2022 Mar 25;13(4):581. doi: 10.3390/genes13040581. Genes (Basel). 2022. PMID: 35456387 Free PMC article. Review.
References
-
- Marini J.C., Forlino A., Cabral W.A., Barnes A.M., San Antonio J.D., Milgrom S.. et al. (2007) Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 28, 209–22110.1002/humu.20429 - DOI - PMC - PubMed
-
- CalzavaraPinton P. and Ritelli M. (2017) Delineation of Ehlers–Danlos syndrome phenotype due to the mutation (c.934C>T, p.Arg312Cys) in COL1A1: report on a three-generation family without cardiovascular events, and literature review. Am. J. Med. Genet. 173, 524–530 - PubMed
-
- Byers P.H., Duvic M., Atkinson M., Robinow M., Smith L.T., Krane S.M.. et al. (1997) Ehlers–Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am. J. Med. Genet. 72, 94–10510.1002/(SICI)1096-8628(19971003)72:1<94::AID-AJMG20>3.0.CO;2-O - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous