

redbiom: a Rapid Sample Discovery and Feature Characterization System
- PMID: 31239397
- PMCID: PMC6593222
- DOI: 10.1128/mSystems.00215-19
redbiom: a Rapid Sample Discovery and Feature Characterization System
Abstract
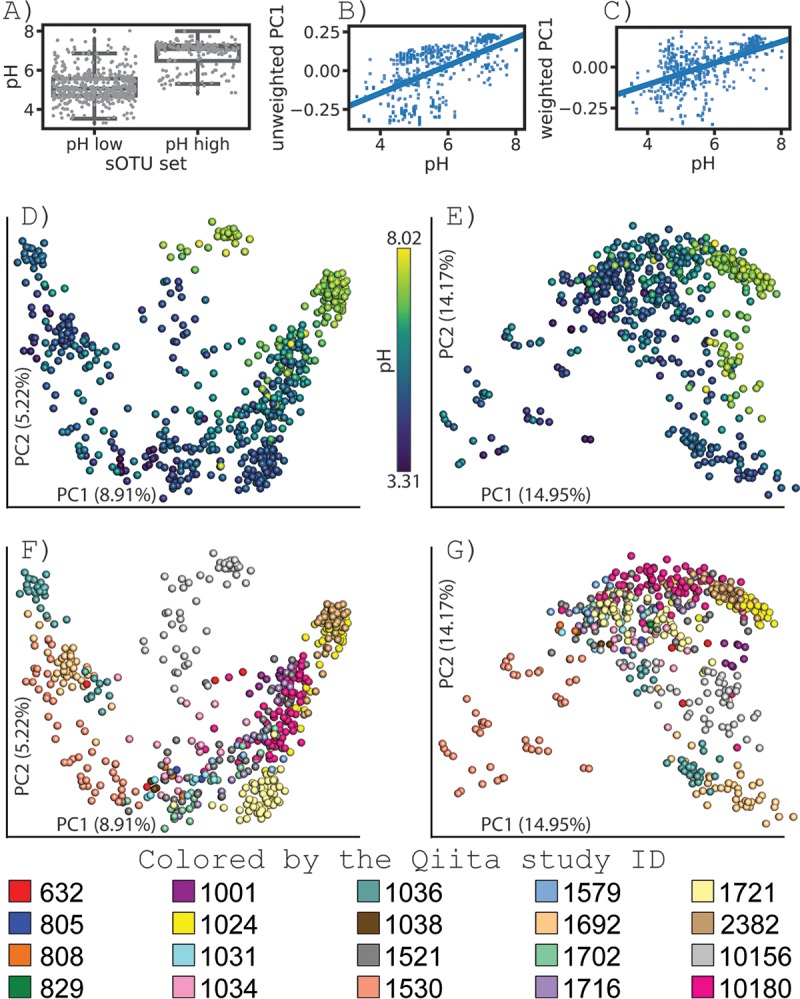
Meta-analyses at the whole-community level have been important in microbiome studies, revealing profound features that structure Earth's microbial communities, such as the unique differentiation of microbes from the mammalian gut relative to free-living microbial communities, the separation of microbiomes in saline and nonsaline environments, and the role of pH in driving soil microbial compositions. However, our ability to identify the specific features of a microbiome that differentiate these community-level patterns have lagged behind, especially as ever-cheaper DNA sequencing has yielded increasingly large data sets. One critical gap is the ability to search for samples that contain specific features (for example, sub-operational taxonomic units [sOTUs] identified by high-resolution statistical methods for removing amplicon sequencing errors). Here we introduce redbiom, a microbiome caching layer, which allows users to rapidly query samples that contain a given feature, retrieve sample data and metadata, and search for samples that match specified metadata values or ranges (e.g., all samples with a pH of >7), implemented using an in-memory NoSQL database called Redis. By default, redbiom allows public anonymous sample access for over 100,000 publicly available samples in the Qiita database. At over 100,000 samples, the caching server requires only 35 GB of resident memory. We highlight how redbiom enables a new type of characterization of microbiome samples and provide tutorials for using redbiom with QIIME 2. redbiom is open source under the BSD license, hosted on GitHub, and can be deployed independently of Qiita to enable search of proprietary or clinically restricted microbiome databases.IMPORTANCE Although analyses that combine many microbiomes at the whole-community level have become routine, searching rapidly for microbiomes that contain a particular sequence has remained difficult. The software we present here, redbiom, dramatically accelerates this process, allowing samples that contain microbiome features to be rapidly identified. This is especially useful when taxonomic annotation is limited, allowing users to identify environments in which unannotated microbes of interest were previously observed. This approach also allows environmental or clinical factors that correlate with specific features, or vice versa, to be identified rapidly, even at a scale of billions of sequences in hundreds of thousands of samples. The software is integrated with existing analysis tools to enable fast, large-scale microbiome searches and discovery of new microbiome relationships.
Keywords: database; meta-analysis; microbiome.
Copyright © 2019 McDonald et al.
Figures

Similar articles
-
Microbiome Search Engine 2: a Platform for Taxonomic and Functional Search of Global Microbiomes on the Whole-Microbiome Level.mSystems. 2021 Jan 19;6(1):e00943-20. doi: 10.1128/mSystems.00943-20. mSystems. 2021. PMID: 33468706 Free PMC article.
-
Calour: an Interactive, Microbe-Centric Analysis Tool.mSystems. 2019 Jan 29;4(1):e00269-18. doi: 10.1128/mSystems.00269-18. eCollection 2019 Jan-Feb. mSystems. 2019. PMID: 30701193 Free PMC article.
-
Identifying and Predicting Novelty in Microbiome Studies.mBio. 2018 Nov 13;9(6):e02099-18. doi: 10.1128/mBio.02099-18. mBio. 2018. PMID: 30425147 Free PMC article.
-
Big Data for a Small World: A Review on Databases and Resources for Studying Microbiomes.J Indian Inst Sci. 2023 Apr 5:1-17. doi: 10.1007/s41745-023-00370-z. Online ahead of print. J Indian Inst Sci. 2023. PMID: 37362854 Free PMC article. Review.
-
Personalized microbiome dynamics - Cytometric fingerprints for routine diagnostics.Mol Aspects Med. 2018 Feb;59:123-134. doi: 10.1016/j.mam.2017.06.005. Epub 2017 Jul 8. Mol Aspects Med. 2018. PMID: 28669592 Review.
Cited by
-
Method development for cross-study microbiome data mining: Challenges and opportunities.Comput Struct Biotechnol J. 2020 Aug 1;18:2075-2080. doi: 10.1016/j.csbj.2020.07.020. eCollection 2020. Comput Struct Biotechnol J. 2020. PMID: 32802279 Free PMC article. Review.
-
Optimizing UniFrac with OpenACC Yields Greater Than One Thousand Times Speed Increase.mSystems. 2022 Jun 28;7(3):e0002822. doi: 10.1128/msystems.00028-22. Epub 2022 May 31. mSystems. 2022. PMID: 35638356 Free PMC article.
-
Efficient computation of Faith's phylogenetic diversity with applications in characterizing microbiomes.Genome Res. 2021 Nov;31(11):2131-2137. doi: 10.1101/gr.275777.121. Epub 2021 Sep 3. Genome Res. 2021. PMID: 34479875 Free PMC article.
-
Consumption of Fermented Foods Is Associated with Systematic Differences in the Gut Microbiome and Metabolome.mSystems. 2020 Mar 17;5(2):e00901-19. doi: 10.1128/mSystems.00901-19. mSystems. 2020. PMID: 32184365 Free PMC article.
-
Human Skin, Oral, and Gut Microbiomes Predict Chronological Age.mSystems. 2020 Feb 11;5(1):e00630-19. doi: 10.1128/mSystems.00630-19. mSystems. 2020. PMID: 32047061 Free PMC article.
References
-
- Sinha R, Abu-Ali G, Vogtmann E, Fodor AA, Ren B, Amir A, Schwager E, Crabtree J, Ma S, Microbiome Quality Control Project Consortium, Abnet CC, Knight R, White O, Huttenhower C. 2017. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat Biotechnol 35:1077–1086. doi:10.1038/nbt.3981. - DOI - PMC - PubMed
-
- Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, Navas-Molina JA, Janssen S, Kopylova E, Vázquez-Baeza Y, González A, Morton JT, Mirarab S, Zech Xu Z, Jiang L, Haroon MF, Kanbar J, Zhu Q, Jin Song S, Kosciolek T, Bokulich NA, Lefler J, Brislawn CJ, Humphrey G, Owens SM, Hampton-Marcell J, Berg-Lyons D, McKenzie V, Fierer N, Fuhrman JA, Clauset A, Stevens RL, Shade A, Pollard KS, Goodwin KD, Jansson JK, Gilbert JA, Knight R, Earth Microbiome Project Consortium. 2017. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551:457–463. doi:10.1038/nature24621. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
