

Common and distinct transcriptional signatures of mammalian embryonic lethality
- PMID: 31243271
- PMCID: PMC6594971
- DOI: 10.1038/s41467-019-10642-x
Common and distinct transcriptional signatures of mammalian embryonic lethality
Abstract
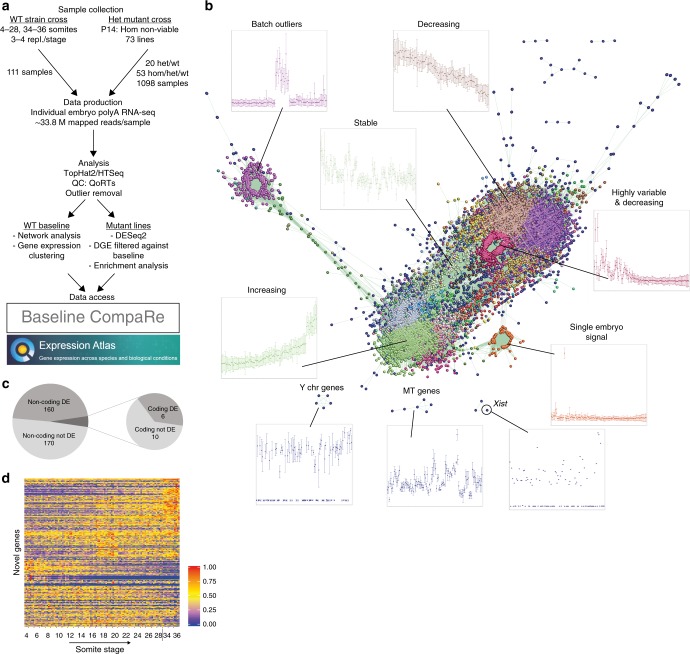
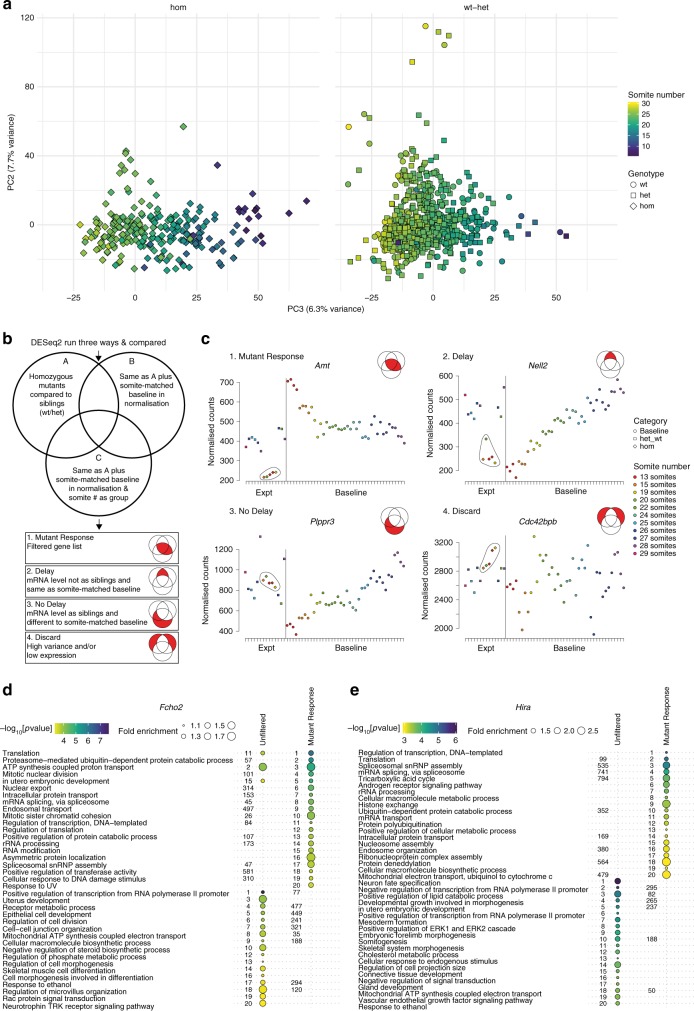
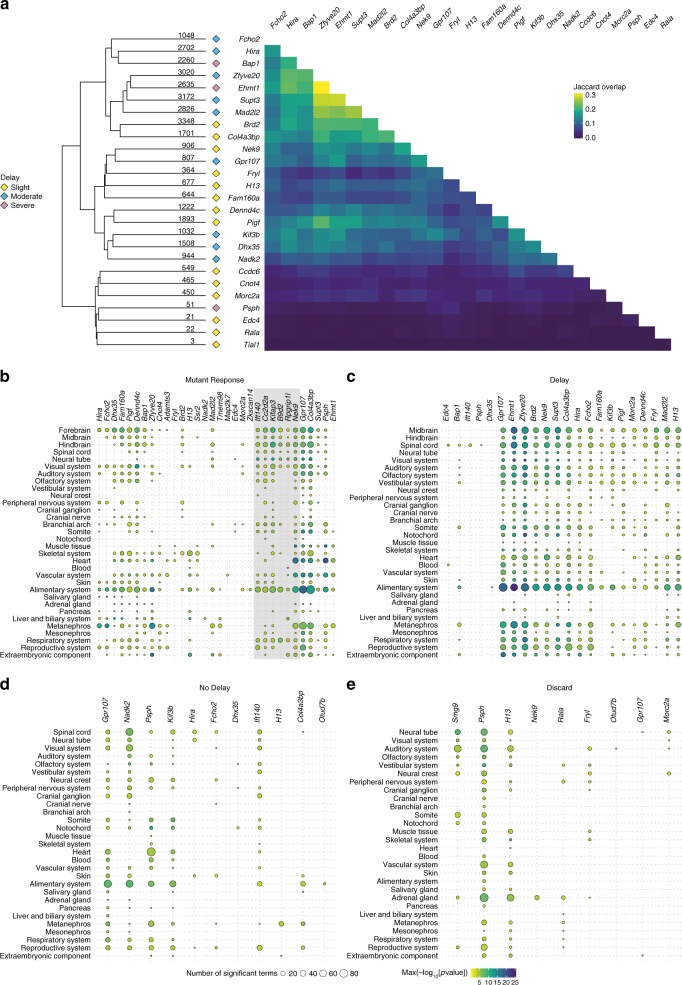
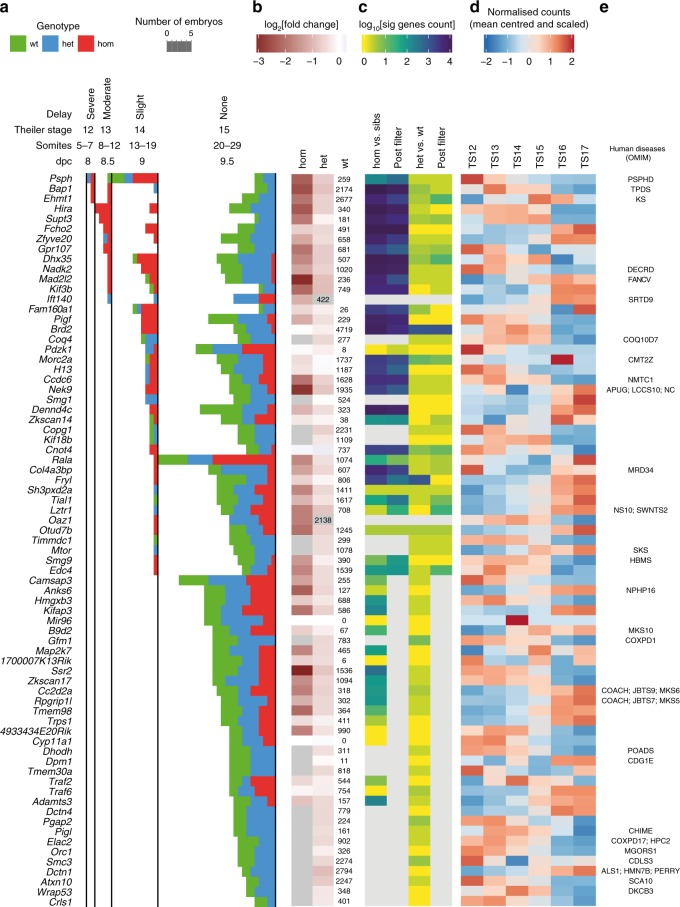
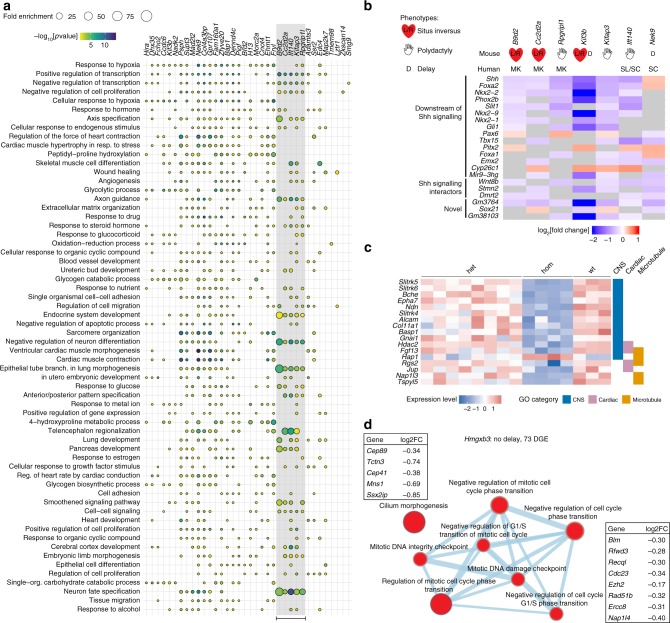
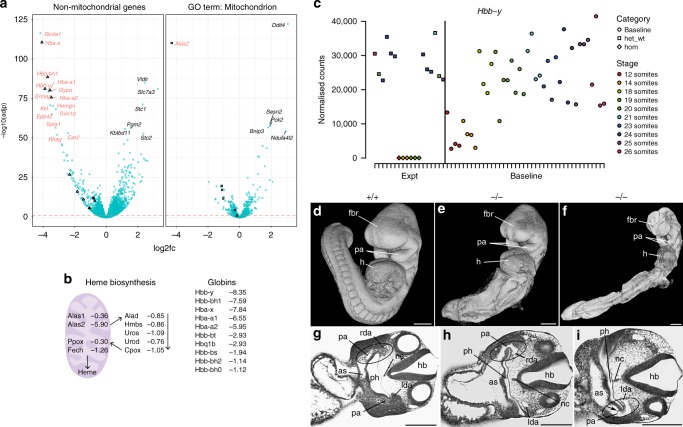
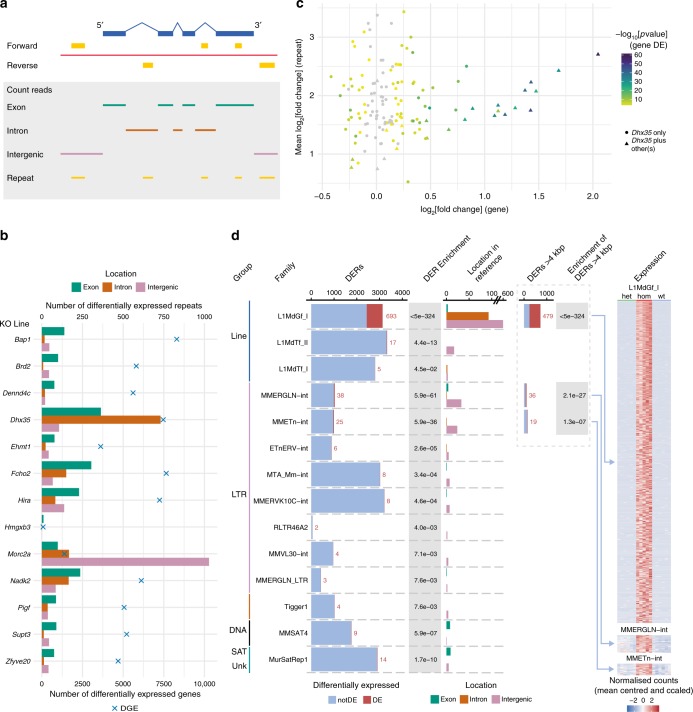
The Deciphering the Mechanisms of Developmental Disorders programme has analysed the morphological and molecular phenotypes of embryonic and perinatal lethal mouse mutant lines in order to investigate the causes of embryonic lethality. Here we show that individual whole-embryo RNA-seq of 73 mouse mutant lines (>1000 transcriptomes) identifies transcriptional events underlying embryonic lethality and associates previously uncharacterised genes with specific pathways and tissues. For example, our data suggest that Hmgxb3 is involved in DNA-damage repair and cell-cycle regulation. Further, we separate embryonic delay signatures from mutant line-specific transcriptional changes by developing a baseline mRNA expression catalogue of wild-type mice during early embryogenesis (4-36 somites). Analysis of transcription outside coding sequence identifies deregulation of repetitive elements in Morc2a mutants and a gene involved in gene-specific splicing. Collectively, this work provides a large scale resource to further our understanding of early embryonic developmental disorders.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
