

Diverse hydrogen production and consumption pathways influence methane production in ruminants
- PMID: 31243332
- PMCID: PMC6776011
- DOI: 10.1038/s41396-019-0464-2
Diverse hydrogen production and consumption pathways influence methane production in ruminants
Abstract
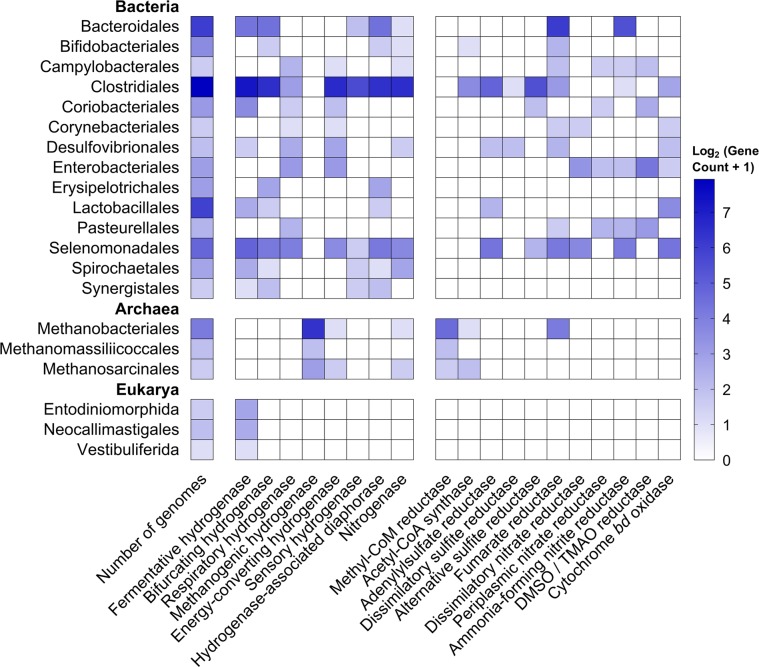
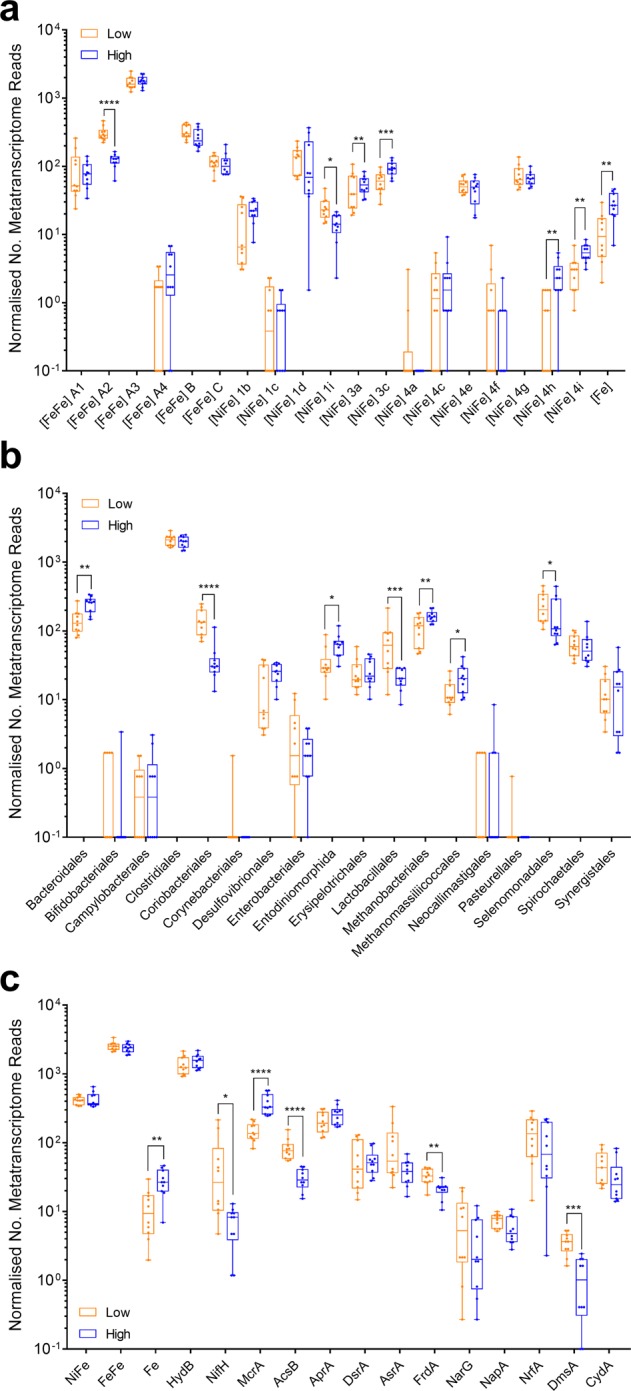
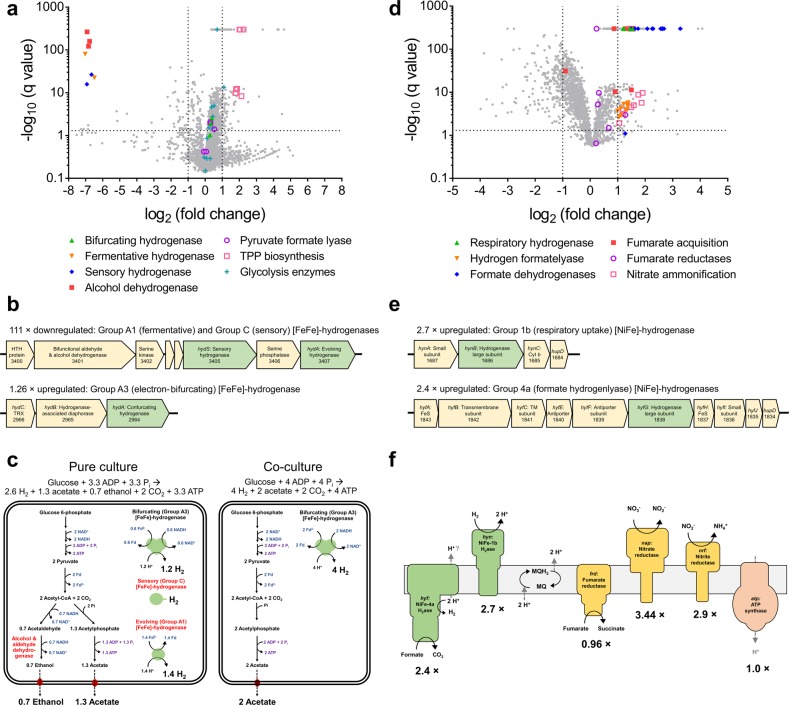
Farmed ruminants are the largest source of anthropogenic methane emissions globally. The methanogenic archaea responsible for these emissions use molecular hydrogen (H2), produced during bacterial and eukaryotic carbohydrate fermentation, as their primary energy source. In this work, we used comparative genomic, metatranscriptomic and co-culture-based approaches to gain a system-wide understanding of the organisms and pathways responsible for ruminal H2 metabolism. Two-thirds of sequenced rumen bacterial and archaeal genomes encode enzymes that catalyse H2 production or consumption, including 26 distinct hydrogenase subgroups. Metatranscriptomic analysis confirmed that these hydrogenases are differentially expressed in sheep rumen. Electron-bifurcating [FeFe]-hydrogenases from carbohydrate-fermenting Clostridia (e.g., Ruminococcus) accounted for half of all hydrogenase transcripts. Various H2 uptake pathways were also expressed, including methanogenesis (Methanobrevibacter), fumarate and nitrite reduction (Selenomonas), and acetogenesis (Blautia). Whereas methanogenesis-related transcripts predominated in high methane yield sheep, alternative uptake pathways were significantly upregulated in low methane yield sheep. Complementing these findings, we observed significant differential expression and activity of the hydrogenases of the hydrogenogenic cellulose fermenter Ruminococcus albus and the hydrogenotrophic fumarate reducer Wolinella succinogenes in co-culture compared with pure culture. We conclude that H2 metabolism is a more complex and widespread trait among rumen microorganisms than previously recognised. There is evidence that alternative hydrogenotrophs, including acetogenic and respiratory bacteria, can prosper in the rumen and effectively compete with methanogens for H2. These findings may help to inform ongoing strategies to mitigate methane emissions by increasing flux through alternative H2 uptake pathways, including through animal selection, dietary supplementation and methanogenesis inhibitors.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
-
- Kirschke S, Bousquet P, Ciais P, Saunois M, Canadell JG, Dlugokencky EJ, et al. Three decades of global methane sources and sinks. Nat Geosci. 2013;6:813–23. doi: 10.1038/ngeo1955. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
