

Analysis of RNA Methylation by Phylogenetically Diverse Cfr Radical S-Adenosylmethionine Enzymes Reveals an Iron-Binding Accessory Domain in a Clostridial Enzyme
- PMID: 31246421
- PMCID: PMC6800567
- DOI: 10.1021/acs.biochem.9b00197
Analysis of RNA Methylation by Phylogenetically Diverse Cfr Radical S-Adenosylmethionine Enzymes Reveals an Iron-Binding Accessory Domain in a Clostridial Enzyme
Abstract
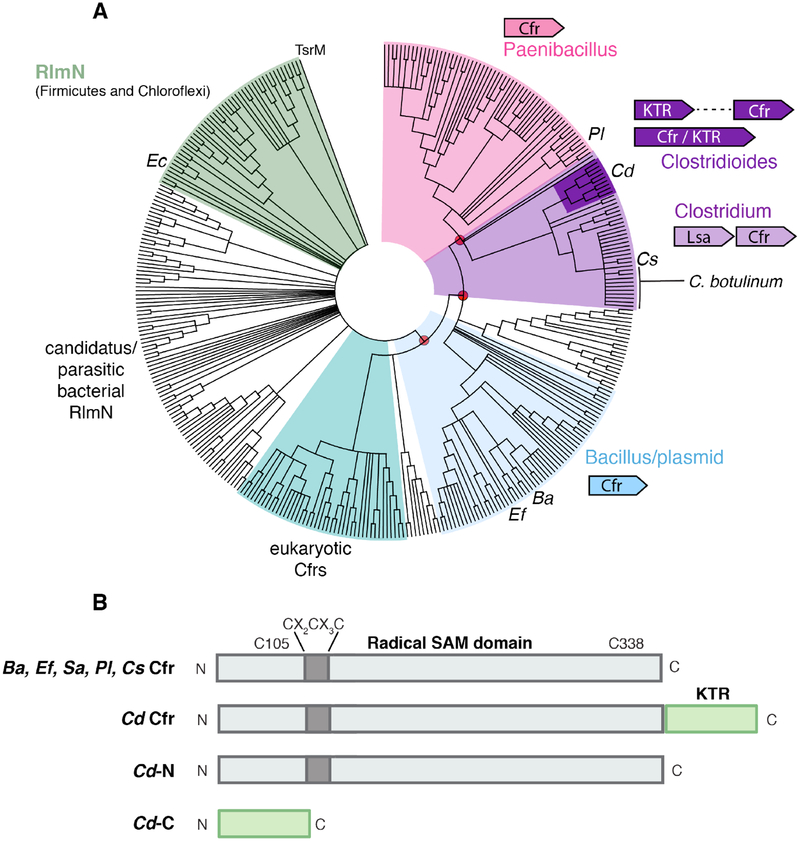
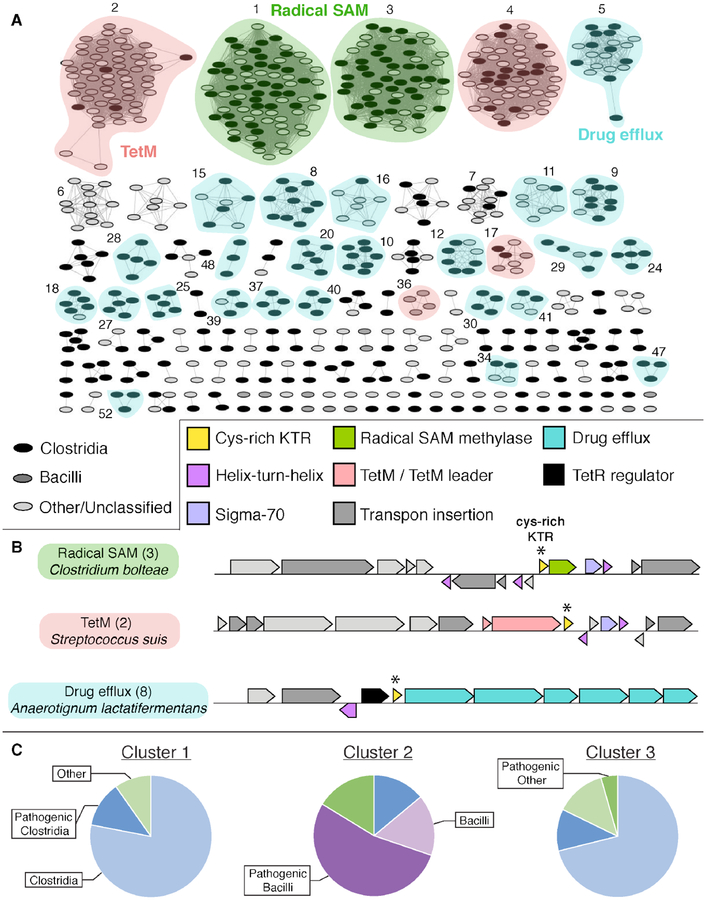
Cfr is a radical S-adenosylmethionine (SAM) RNA methylase linked to multidrug antibiotic resistance in bacterial pathogens. It catalyzes a chemically challenging C-C bond-forming reaction to methylate C8 of A2503 (Escherichia coli numbering) of 23S rRNA during ribosome assembly. The cfr gene has been identified as a mobile genetic element in diverse bacteria and in the genome of select Bacillales and Clostridiales species. Despite the importance of Cfr, few representatives have been purified and characterized in vitro. Here we show that Cfr homologues from Bacillus amyloliquefaciens, Enterococcus faecalis, Paenibacillus lautus, and Clostridioides difficile act as C8 adenine RNA methylases in biochemical assays. C. difficile Cfr contains an additional Cys-rich C-terminal domain that binds a mononuclear Fe2+ ion in a rubredoxin-type Cys4 motif. The C-terminal domain can be truncated with minimal impact on C. difficile Cfr activity, but the rate of turnover is decreased upon disruption of the Fe2+-binding site by Zn2+ substitution or ligand mutation. These findings indicate an important purpose for the observed C-terminal iron in the native fusion protein. Bioinformatic analysis of the C. difficile Cfr Cys-rich domain shows that it is widespread (∼1400 homologues) as a stand-alone gene in pathogenic or commensal Bacilli and Clostridia, with >10% encoded adjacent to a predicted radical SAM RNA methylase. Although the domain is not essential for in vitro C. difficile Cfr activity, the genomic co-occurrence and high abundance in the human microbiome suggest a possible functional role for a specialized rubredoxin in certain radical SAM RNA methylases that are relevant to human health.
Figures







References
-
- Finn RD, Attwood TK, Babbitt PC, Bateman A, Bork P, Bridge AJ, Chang HY, Dosztanyi Z, El-Gebali S, Fraser M, Gough J, Haft D, Holliday GL, Huang H, Huang X, Letunic I, Lopez R, Lu S, Marchler-Bauer A, Mi H, Mistry J, Natale DA, Necci M, Nuka G, Orengo CA, Park Y, Pesseat S, Piovesan D, Potter SC, Rawlings ND, Redaschi N, Richardson L, Rivoire C, Sangrador-Vegas A, Sigrist C, Sillitoe I, Smithers B, Squizzato S, Sutton G, Thanki N, Thomas PD, Tosatto SC, Wu CH, Xenarios I, Yeh LS, Young SY, and Mitchell AL (2017) InterPro in 2017-beyond protein family and domain annotations, Nucleic Acids Res 45, D190–D199. - PMC - PubMed
-
- Frey PA, Hegeman AD, and Ruzicka FJ (2008) The radical SAM superfamily, Crit Rev Biochem Mol Biol 43, 63–88. - PubMed
-
- Landgraf BJ, McCarthy EL, and Booker SJ (2016) Radical S-adenosylmethionine enzymes in human health and disease, Annu Rev Biochem 85, 485–514. - PubMed
-
- Kimura S, and Suzuki T (2015) Iron-sulfur proteins responsible for RNA modifications, Biochim Biophys Acta 1853, 1272–1283. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
