

Sensory neuropathy and nociception in rodent models of Parkinson's disease
- PMID: 31248900
- PMCID: PMC6602317
- DOI: 10.1242/dmm.039396
Sensory neuropathy and nociception in rodent models of Parkinson's disease
Abstract
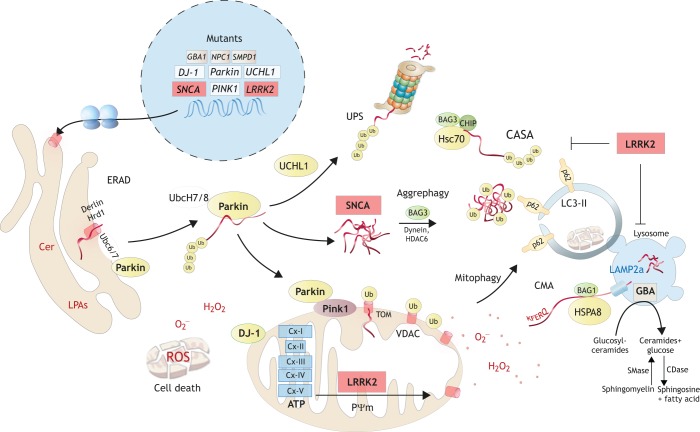
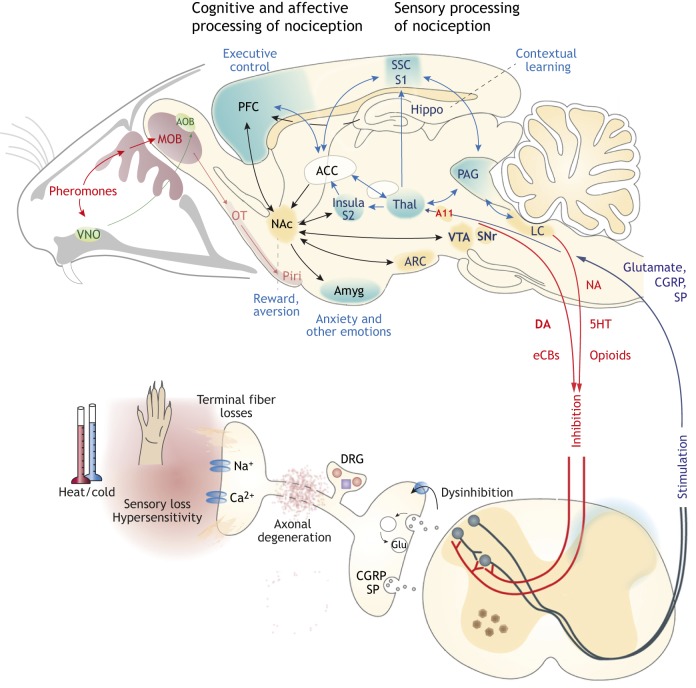
Parkinson's disease (PD) often manifests with prodromal pain and sensory losses whose etiologies are not well understood. Multiple genetic and toxicity-based rodent models of PD partly recapitulate the histopathology and motor function deficits. Although far less studied, there is some evidence that rodents, similar to humans, develop sensory manifestations of the disease, which may precede motor disturbances and help to elucidate the underlying mechanisms of PD-associated pain at the molecular and neuron circuit levels. The present Review summarizes nociception and other sensory functions in frequently used rodent PD models within the context of the complex phenotypes. In terms of mechanisms, it appears that the acute loss of dopaminergic neurons in systemic toxicity models (MPTP, rotenone) primarily causes nociceptive hyperexcitability, presumably owing to a loss of inhibitory control, whereas genetic models primarily result in a progressive loss of heat perception, reflecting sensory fiber neuropathies. At the molecular level, neither α-synuclein deposits alone nor failure of mitophagy alone appear to be strong enough to result in axonal or synaptic pathology of nociceptive neurons that manifest at the behavioral level, and peripheral sensory loss may mask central 'pain' in behavioral tests. Hence, allostatic combinations or additional challenges and novel behavioral assessments are needed to better evaluate PD-associated sensory neuropathies and pain in rodents.
Keywords: Mitogenesis; Mitophagy; Non-motor Parkinson's disease; Pain; Protein aggregate; Sensory neuropathy; Synuclein.
© 2019. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interestsThe authors declare no competing or financial interests.
Figures
Similar articles
-
Electroacupuncture Promotes Recovery of Motor Function and Reduces Dopaminergic Neuron Degeneration in Rodent Models of Parkinson's Disease.Int J Mol Sci. 2017 Aug 24;18(9):1846. doi: 10.3390/ijms18091846. Int J Mol Sci. 2017. PMID: 28837077 Free PMC article.
-
How can rAAV-α-synuclein and the fibril α-synuclein models advance our understanding of Parkinson's disease?J Neurochem. 2016 Oct;139 Suppl 1(Suppl 1):131-155. doi: 10.1111/jnc.13627. Epub 2016 May 4. J Neurochem. 2016. PMID: 27018978 Free PMC article. Review.
-
Cholesterol contributes to dopamine-neuronal loss in MPTP mouse model of Parkinson's disease: Involvement of mitochondrial dysfunctions and oxidative stress.PLoS One. 2017 Feb 7;12(2):e0171285. doi: 10.1371/journal.pone.0171285. eCollection 2017. PLoS One. 2017. PMID: 28170429 Free PMC article.
-
The intranasal administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a new rodent model to test palliative and neuroprotective agents for Parkinson's disease.Curr Pharm Des. 2011;17(5):489-507. doi: 10.2174/138161211795164095. Curr Pharm Des. 2011. PMID: 21375482 Review.
-
A new Drosophila model to study the interaction between genetic and environmental factors in Parkinson's disease.Brain Res. 2014 Oct 2;1583:277-86. doi: 10.1016/j.brainres.2014.08.021. Epub 2014 Aug 15. Brain Res. 2014. PMID: 25130663
Cited by
-
Pain in monogenic Parkinson's disease: a comprehensive review.Front Neurol. 2023 Oct 30;14:1248828. doi: 10.3389/fneur.2023.1248828. eCollection 2023. Front Neurol. 2023. PMID: 38020640 Free PMC article. Review.
-
The Role of AlphαSynuclein in Mouse Models of Acute, Inflammatory and Neuropathic Pain.Cells. 2022 Jun 19;11(12):1967. doi: 10.3390/cells11121967. Cells. 2022. PMID: 35741096 Free PMC article.
-
In the Rat Midbrain, SG2NA and DJ-1 have Common Interactome, Including Mitochondrial Electron Transporters that are Comodulated Under Oxidative Stress.Cell Mol Neurobiol. 2023 Oct;43(7):3061-3080. doi: 10.1007/s10571-023-01356-2. Epub 2023 May 10. Cell Mol Neurobiol. 2023. PMID: 37165139 Free PMC article. Review.
-
Serotonergic Neurotransmission System Modulator, Vortioxetine, and Dopaminergic D2/D3 Receptor Agonist, Ropinirole, Attenuate Fibromyalgia-Like Symptoms in Mice.Molecules. 2021 Apr 20;26(8):2398. doi: 10.3390/molecules26082398. Molecules. 2021. PMID: 33924258 Free PMC article.
-
Failure of Diphtheria Toxin Model to Induce Parkinson-Like Behavior in Mice.Int J Mol Sci. 2021 Aug 31;22(17):9496. doi: 10.3390/ijms22179496. Int J Mol Sci. 2021. PMID: 34502404 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
