

Best practices and benchmarks for intact protein analysis for top-down mass spectrometry
- PMID: 31249407
- PMCID: PMC6719561
- DOI: 10.1038/s41592-019-0457-0
Best practices and benchmarks for intact protein analysis for top-down mass spectrometry
Abstract
One gene can give rise to many functionally distinct proteoforms, each of which has a characteristic molecular mass. Top-down mass spectrometry enables the analysis of intact proteins and proteoforms. Here members of the Consortium for Top-Down Proteomics provide a decision tree that guides researchers to robust protocols for mass analysis of intact proteins (antibodies, membrane proteins and others) from mixtures of varying complexity. We also present cross-platform analytical benchmarks using a protein standard sample, to allow users to gauge their proficiency.
Conflict of interest statement
Authors with company affiliations include J.L.L. (Amgen), K.C.A. (Alnylam), R.S. (Biogen), J.J.W. (Bruker), I.D.G.C. (Amgen), P.O.D. (Eastwoods Consulting) and Y.O.T. (Spectroswiss).
Figures
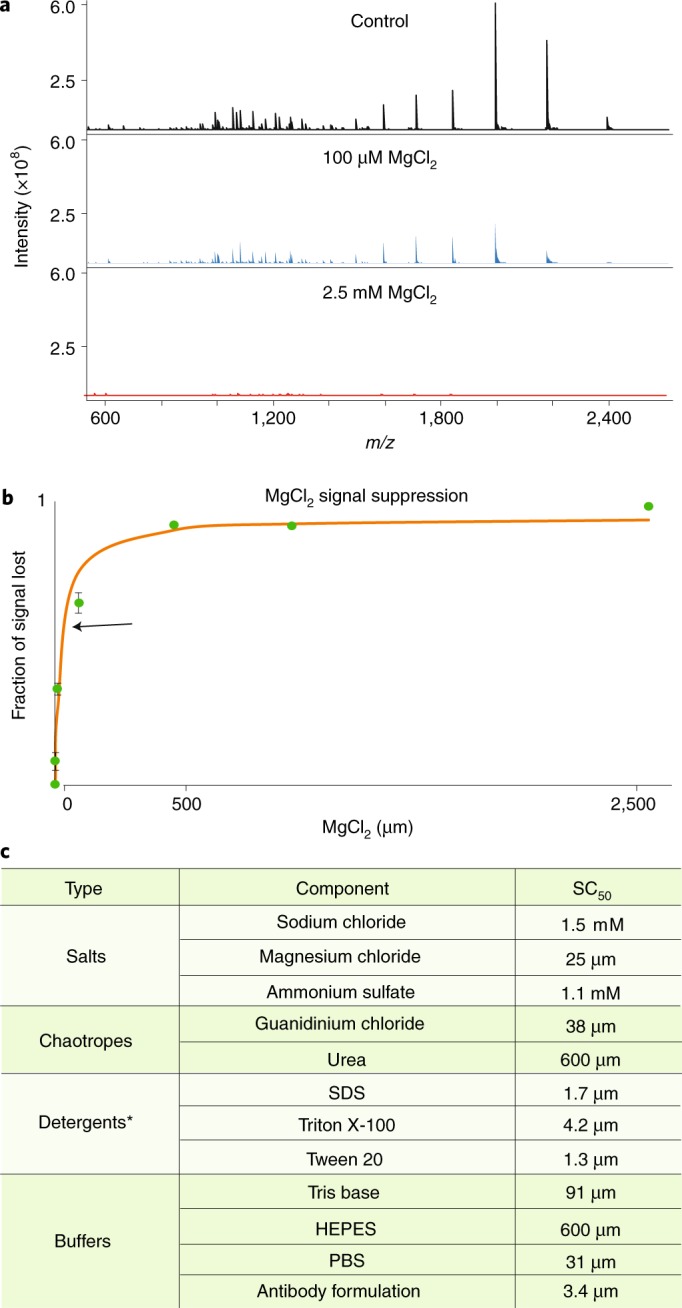
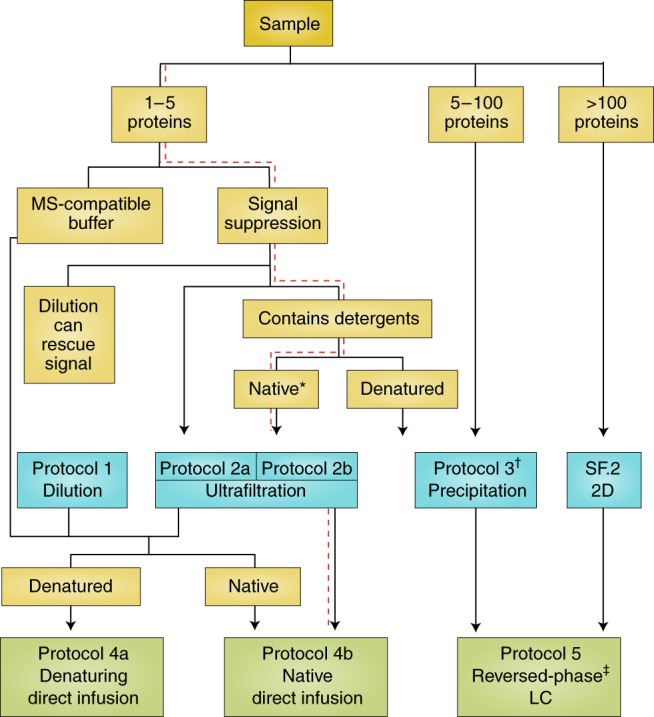



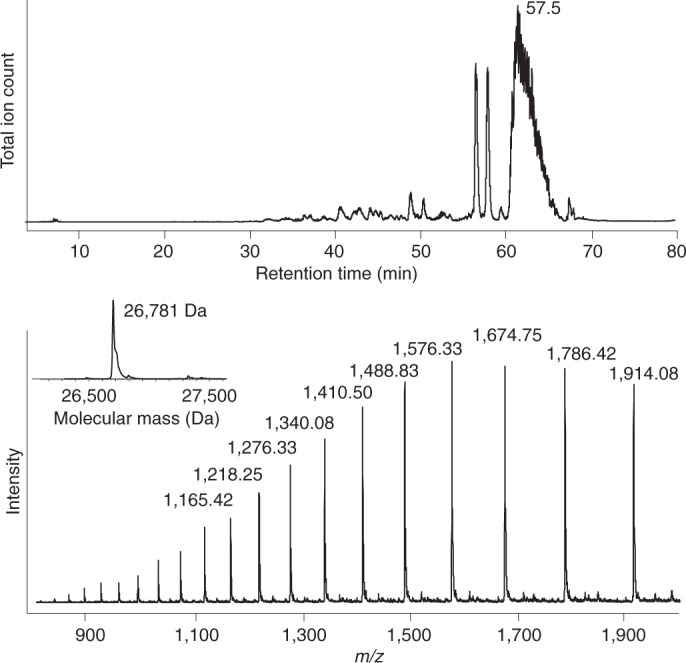
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
