

L-Selectin Enhanced T Cells Improve the Efficacy of Cancer Immunotherapy
- PMID: 31249570
- PMCID: PMC6582763
- DOI: 10.3389/fimmu.2019.01321
L-Selectin Enhanced T Cells Improve the Efficacy of Cancer Immunotherapy
Abstract
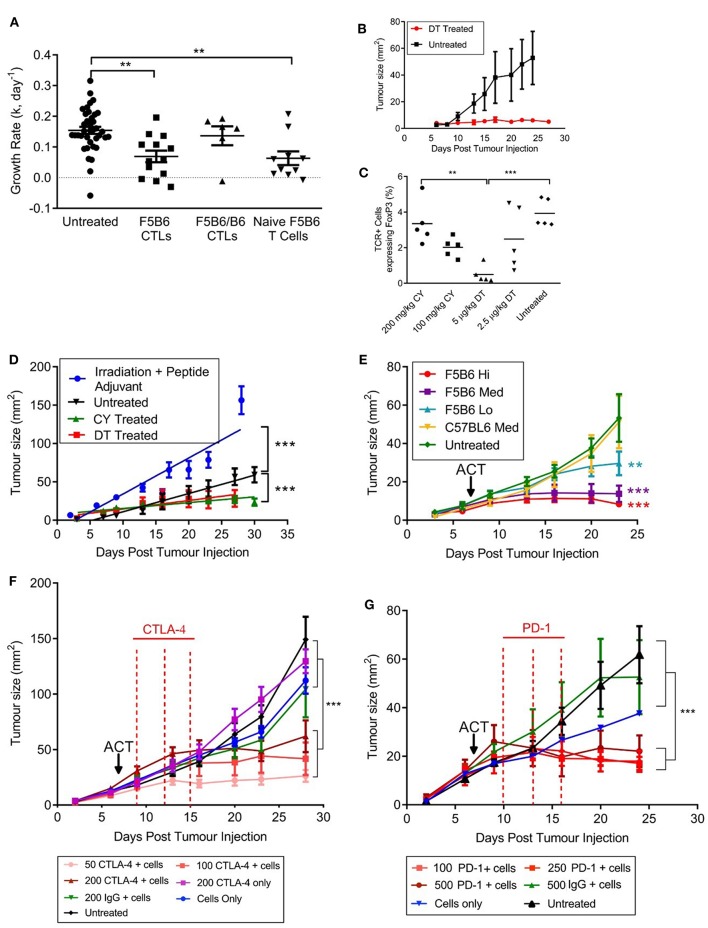
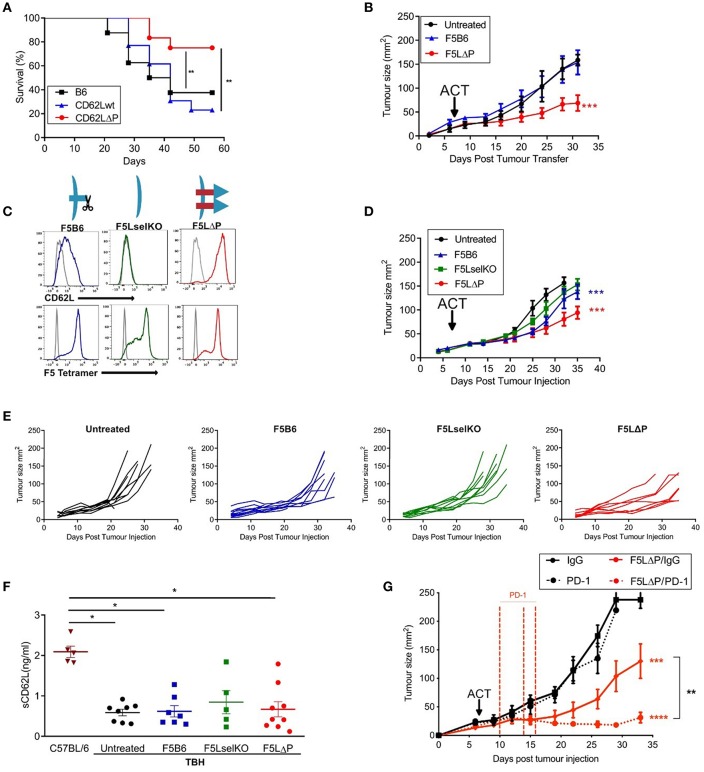
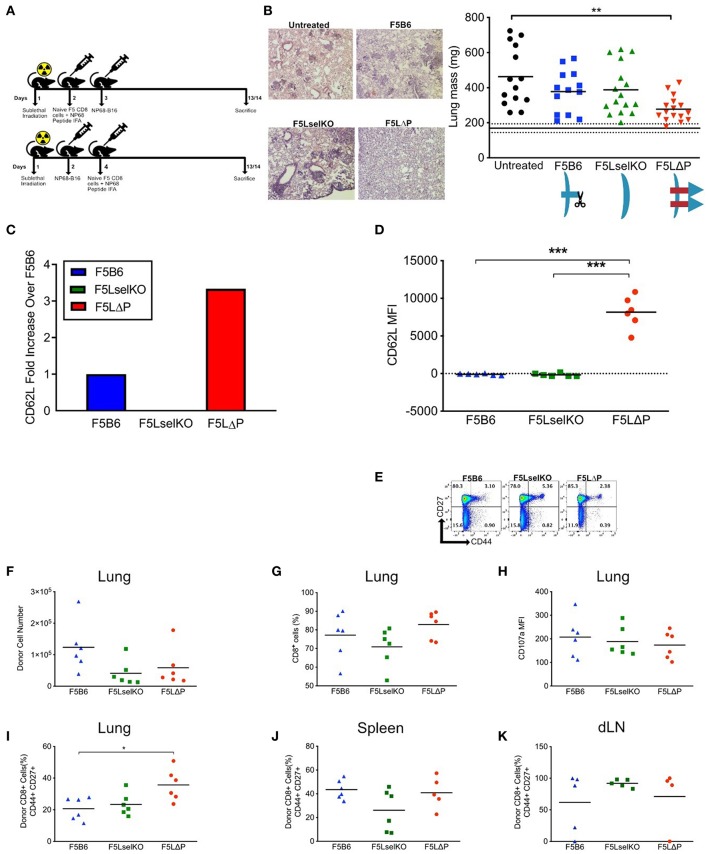
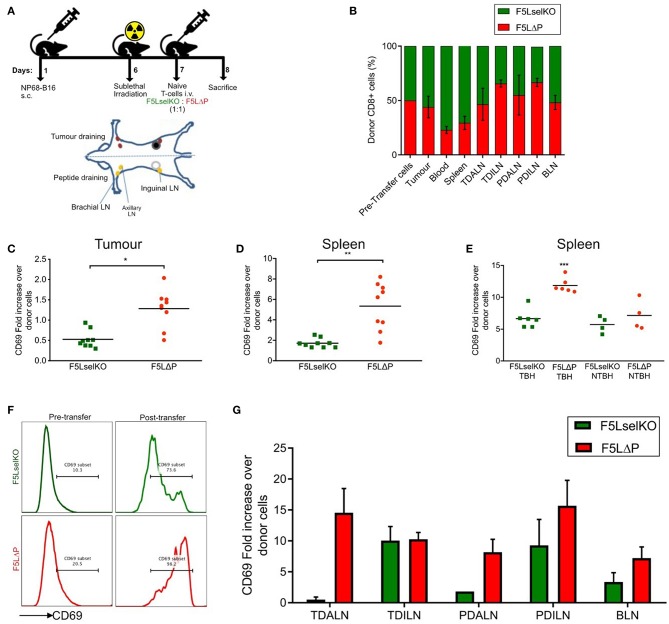
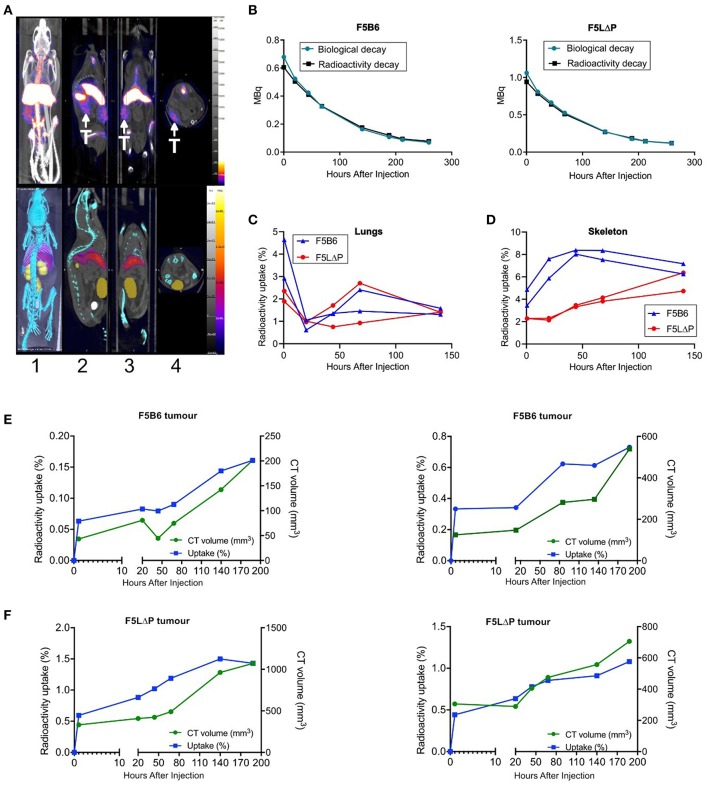
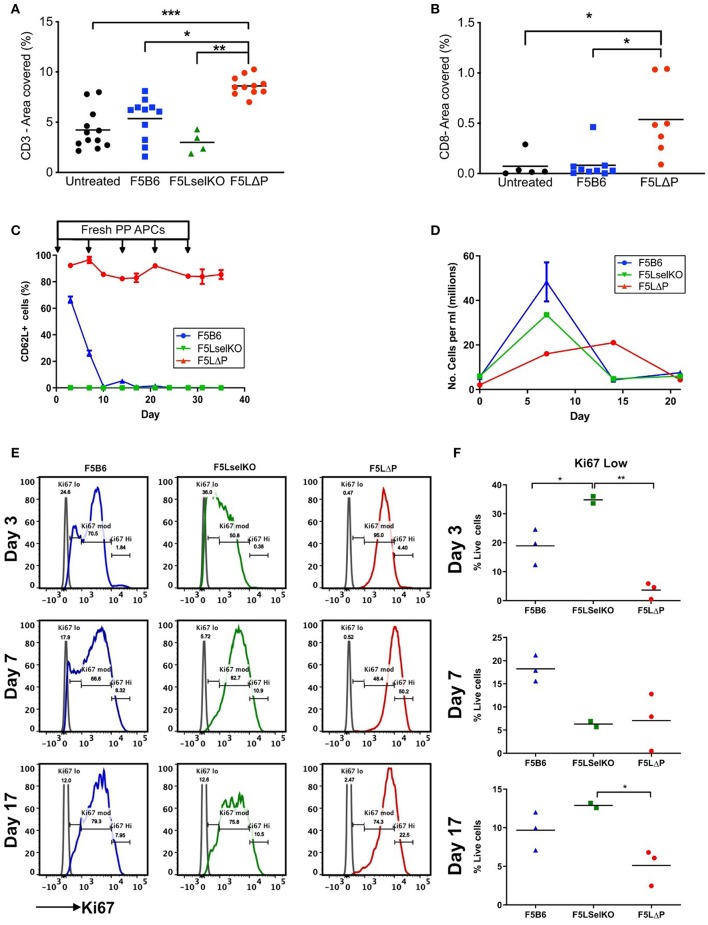
The homing molecule, L-selectin (CD62L), is commonly used as a T cell activation marker, since expression is downregulated following engagement of the T cell receptor. Studies in mice have shown that CD62L+ central memory T cells are better at controlling tumor growth than CD62L- effector memory T cells, while L-selectin knockout T cells are poor at controlling tumor growth. Here, we test the hypothesis that T cells expressing genetically modified forms of L-selectin that are maintained following T cell activation (L-selectin enhanced T cells) are better at controlling tumor growth than wild type T cells. Using mouse models of adoptive cell therapy, we show that L-selectin enhancement improves the efficacy of CD8+ T cells in controlling solid and disseminated tumor growth. L-selectin knockout T cells had no effect. Checkpoint blockade inhibitors synergized with wild type and L-selectin enhanced T cells but had no effect in the absence of T cell transfers. Reduced tumor growth by L-selectin enhanced T cells correlated with increased frequency of CD8+ tumor infiltrating T cells 21 days after commencing therapy. Longitudinal tracking of Zirconium-89 (89Zr) labeled T cells using PET-CT showed that transferred T cells localize to tumors within 1 h and accumulate over the following 7 days. L-selectin did not promote T cell homing to tumors within 18 h of transfer, however the early activation marker CD69 was upregulated on L-selectin positive but not L-selectin knockout T cells. L-selectin positive and L-selectin knockout T cells homed equally well to tumor-draining lymph nodes and spleens. CD69 expression was upregulated on both L-selectin positive and L-selectin knockout T cells but was significantly higher on L-selectin expressing T cells, particularly in the spleen. Clonal expansion of isolated L-selectin enhanced T cells was slower, and L-selectin was linked to expression of proliferation marker Ki67. Together these findings demonstrate that maintaining L-selectin expression on tumor-specific T cells offers an advantage in mouse models of cancer immunotherapy. The beneficial role of L-selectin is unrelated to its' well-known role in T cell homing and, instead, linked to activation of therapeutic T cells inside tumors. These findings suggest that L-selectin may benefit clinical applications in T cell selection for cancer therapy and for modifying CAR-T cells to broaden their clinical scope.
Keywords: L-selectin/CD62L; T cells; adoptive T cell therapy; cancer immunotherapy; melanoma.
Figures
Similar articles
-
Understanding the biology of ex vivo-expanded CD8 T cells for adoptive cell therapy: role of CD62L.Immunol Res. 2013 Dec;57(1-3):23-33. doi: 10.1007/s12026-013-8456-1. Immunol Res. 2013. PMID: 24218360
-
Helper-independent, L-selectinlow CD8+ T cells with broad anti-tumor efficacy are naturally sensitized during tumor progression.J Immunol. 2000 Nov 15;165(10):5738-49. doi: 10.4049/jimmunol.165.10.5738. J Immunol. 2000. PMID: 11067932
-
Adoptive immunotherapy of advanced tumors with CD62 L-selectin(low) tumor-sensitized T lymphocytes following ex vivo hyperexpansion.J Immunol. 2002 Sep 15;169(6):3314-20. doi: 10.4049/jimmunol.169.6.3314. J Immunol. 2002. PMID: 12218152
-
Immunotherapy for advanced melanoma.J Invest Dermatol. 2008 Nov;128(11):2596-2605. doi: 10.1038/jid.2008.101. J Invest Dermatol. 2008. PMID: 18927541 Free PMC article. Review.
-
Adoptive-cell-transfer therapy for the treatment of patients with cancer.Nat Rev Cancer. 2003 Sep;3(9):666-75. doi: 10.1038/nrc1167. Nat Rev Cancer. 2003. PMID: 12951585 Free PMC article. Review.
Cited by
-
The Expression of LDL-R in CD8+ T Cells Serves as an Early Assessment Parameter for the Production of TCR-T Cells.In Vivo. 2023 Nov-Dec;37(6):2480-2489. doi: 10.21873/invivo.13355. In Vivo. 2023. PMID: 37905663 Free PMC article.
-
Cell Adhesion Molecules in Normal Skin and Melanoma.Biomolecules. 2021 Aug 15;11(8):1213. doi: 10.3390/biom11081213. Biomolecules. 2021. PMID: 34439879 Free PMC article. Review.
-
Immune Cell Migration to Cancer.Cells. 2024 May 16;13(10):844. doi: 10.3390/cells13100844. Cells. 2024. PMID: 38786066 Free PMC article. Review.
-
The gene signature of tertiary lymphoid structures within ovarian cancer predicts the prognosis and immunotherapy benefit.Front Genet. 2023 Jan 10;13:1090640. doi: 10.3389/fgene.2022.1090640. eCollection 2022. Front Genet. 2023. PMID: 36704336 Free PMC article.
-
A kit formulation for the preparation of [89Zr]Zr(oxinate)4 for PET cell tracking: White blood cell labelling and comparison with [111In]In(oxinate)3.Nucl Med Biol. 2020 Nov-Dec;90-91:31-40. doi: 10.1016/j.nucmedbio.2020.09.002. Epub 2020 Sep 15. Nucl Med Biol. 2020. PMID: 32979725 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials