

Transcriptome signature of cellular senescence
- PMID: 31251810
- PMCID: PMC6698740
- DOI: 10.1093/nar/gkz555
Transcriptome signature of cellular senescence
Erratum in
-
Transcriptome signature of cellular senescence.Nucleic Acids Res. 2019 Dec 2;47(21):11476. doi: 10.1093/nar/gkz879. Nucleic Acids Res. 2019. PMID: 31612919 Free PMC article. No abstract available.
Abstract
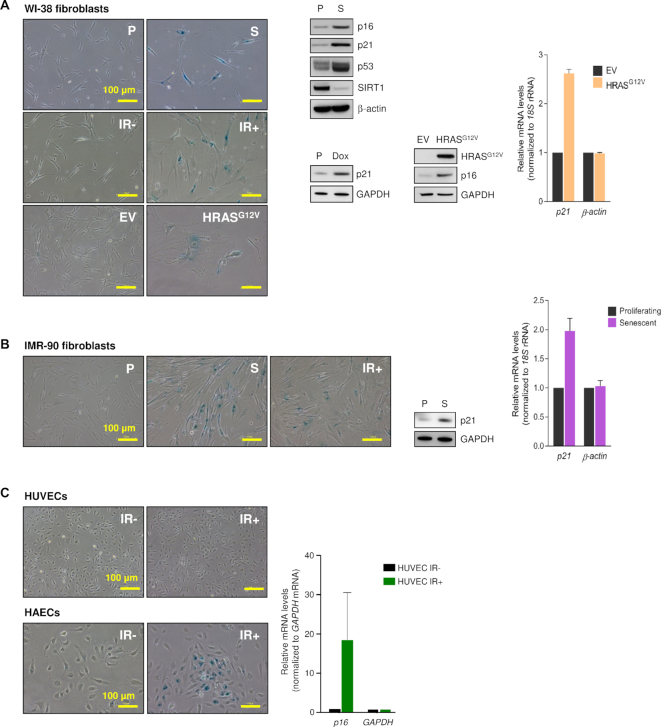
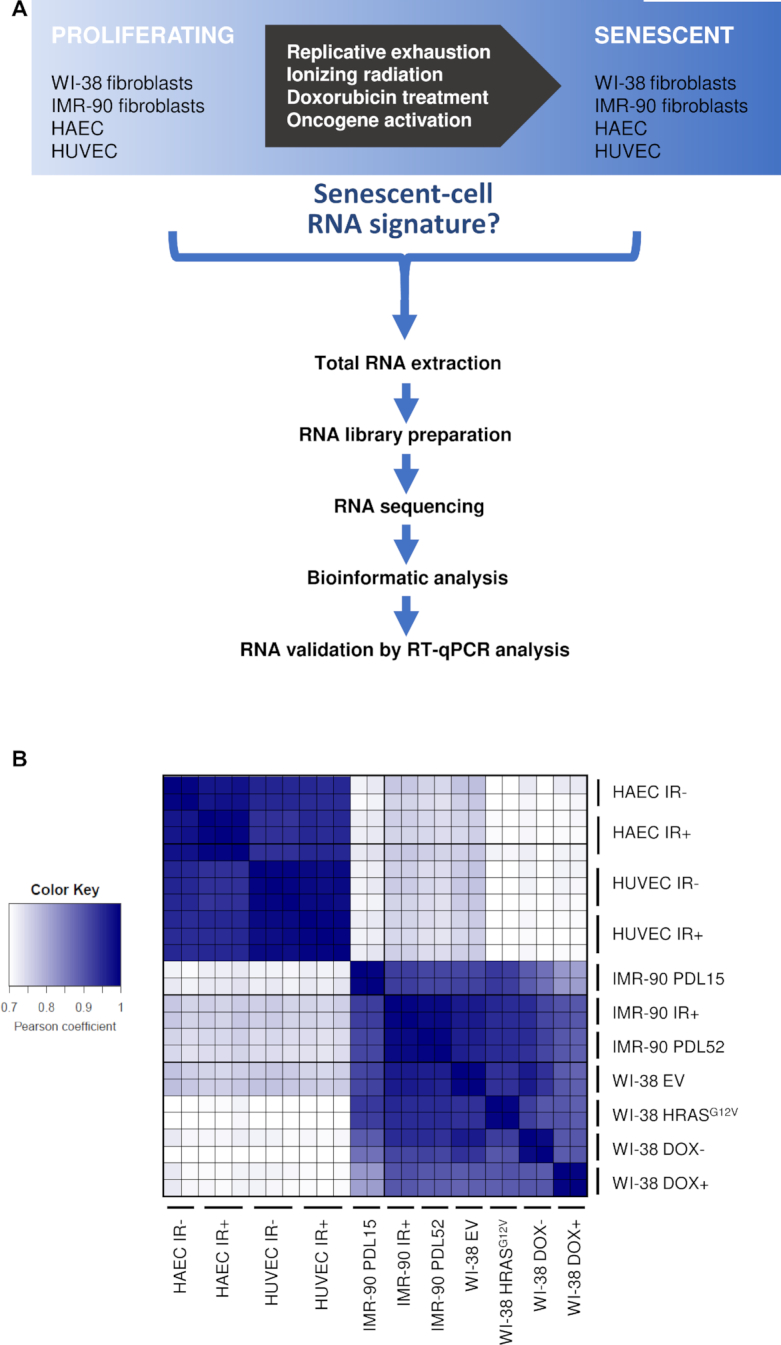
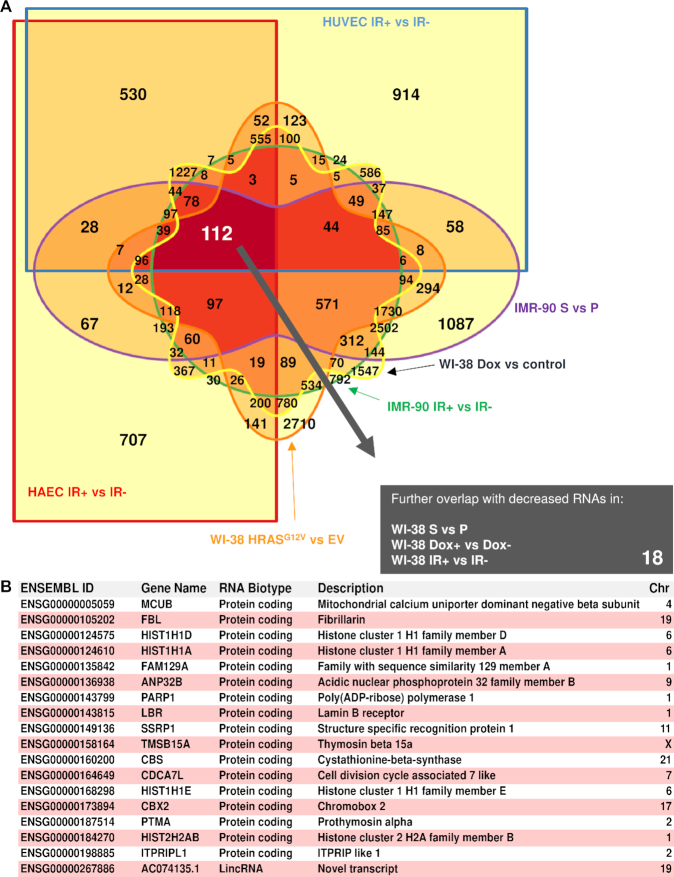
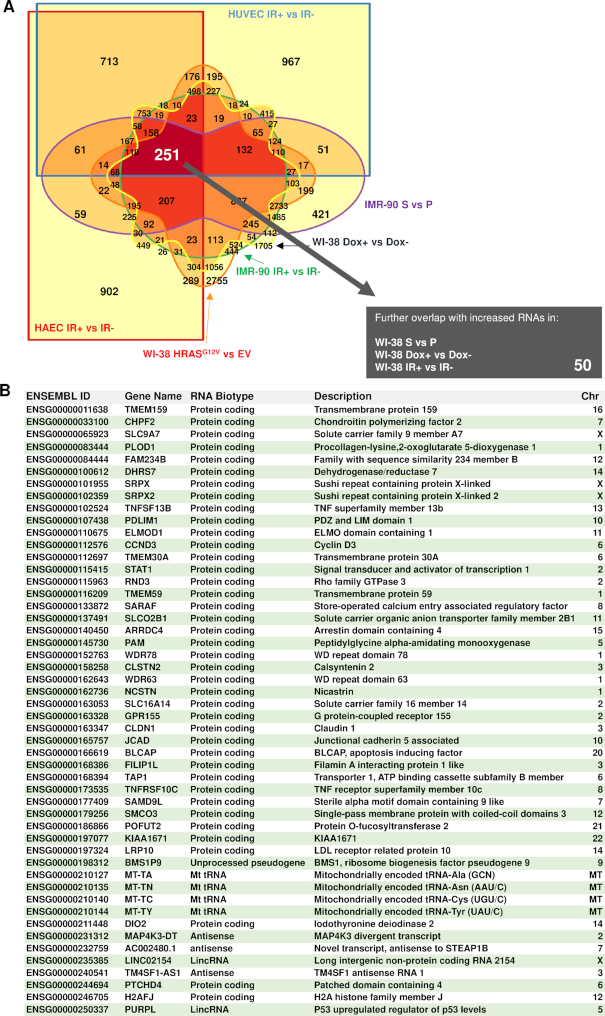
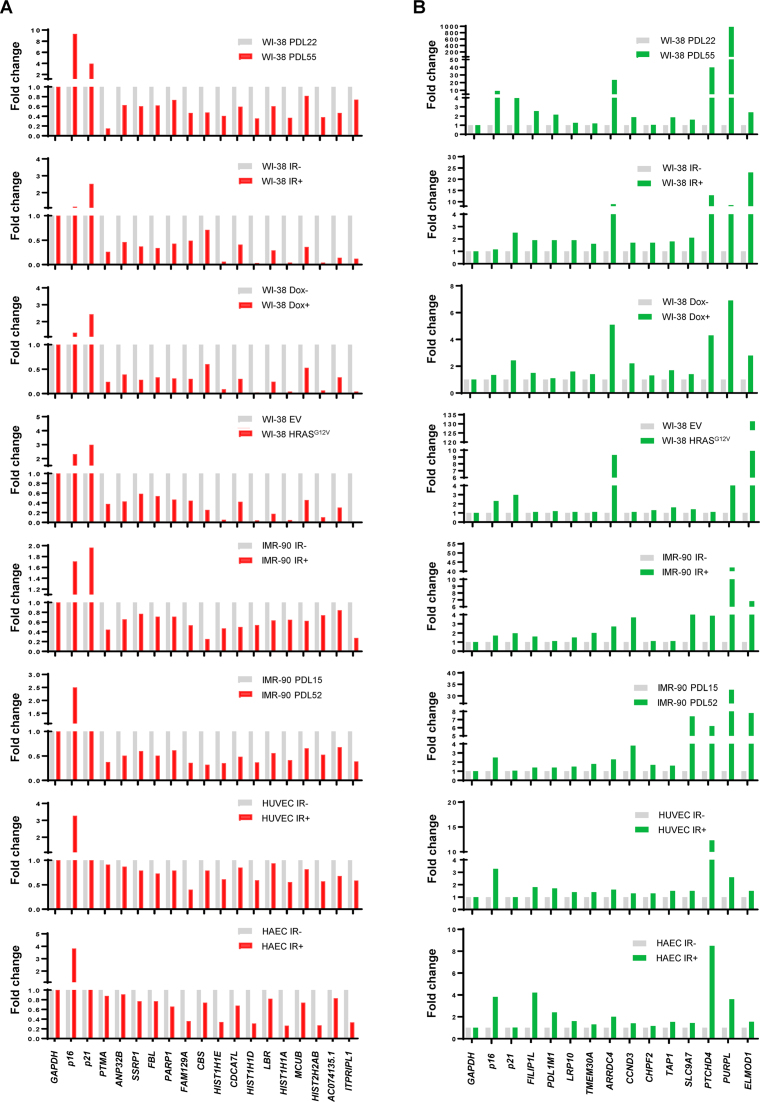
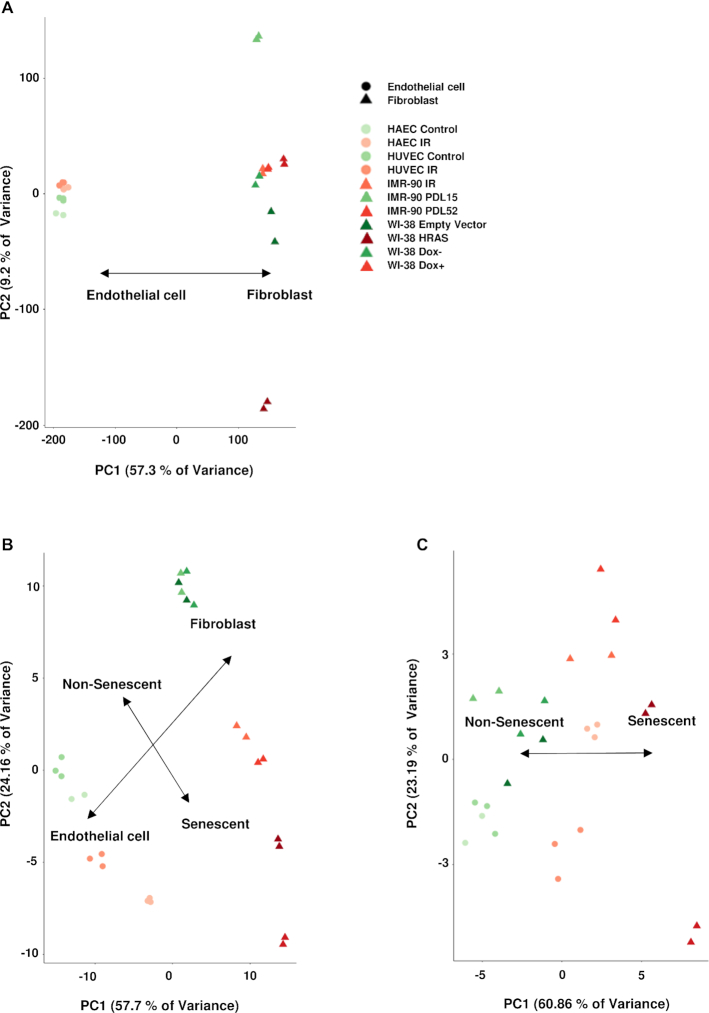
Cellular senescence, an integral component of aging and cancer, arises in response to diverse triggers, including telomere attrition, macromolecular damage and signaling from activated oncogenes. At present, senescent cells are identified by the combined presence of multiple traits, such as senescence-associated protein expression and secretion, DNA damage and β-galactosidase activity; unfortunately, these traits are neither exclusively nor universally present in senescent cells. To identify robust shared markers of senescence, we have performed RNA-sequencing analysis across eight diverse models of senescence triggered in human diploid fibroblasts (WI-38, IMR-90) and endothelial cells (HUVEC, HAEC) by replicative exhaustion, exposure to ionizing radiation or doxorubicin, and expression of the oncogene HRASG12V. The intersection of the altered transcriptomes revealed 50 RNAs consistently elevated and 18 RNAs consistently reduced across all senescence models, including many protein-coding mRNAs and some non-coding RNAs. We propose that these shared transcriptome profiles will enable the identification of senescent cells in vivo, the investigation of their roles in aging and malignancy and the development of strategies to target senescent cells therapeutically.
Published by Oxford University Press on behalf of Nucleic Acids Research 2019.
Figures
References
-
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965; 37:614–636. - PubMed
-
- Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001; 11:S27–S31. - PubMed
-
- Zhang R., Adams P.D.. Heterochromatin and its relationship to cell senescence and cancer therapy. Cell Cycle. 2007; 6:784–789. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
