

Microbiome Signatures Associated With Steatohepatitis and Moderate to Severe Fibrosis in Children With Nonalcoholic Fatty Liver Disease
- PMID: 31255652
- PMCID: PMC6756995
- DOI: 10.1053/j.gastro.2019.06.028
Microbiome Signatures Associated With Steatohepatitis and Moderate to Severe Fibrosis in Children With Nonalcoholic Fatty Liver Disease
Abstract
Background & aims: The intestinal microbiome might affect the development and severity of nonalcoholic fatty liver disease (NAFLD). We analyzed microbiomes of children with and without NAFLD.
Methods: We performed a prospective, observational, cross-sectional study of 87 children (age range, 8-17 years) with biopsy-proven NAFLD and 37 children with obesity without NAFLD (controls). Fecal samples were collected and microbiome composition and functions were assessed using 16S ribosomal RNA amplicon sequencing and metagenomic shotgun sequencing. Microbial taxa were identified using zero-inflated negative binomial modeling. Genes contributing to bacterial pathways were identified using gene set enrichment analysis.
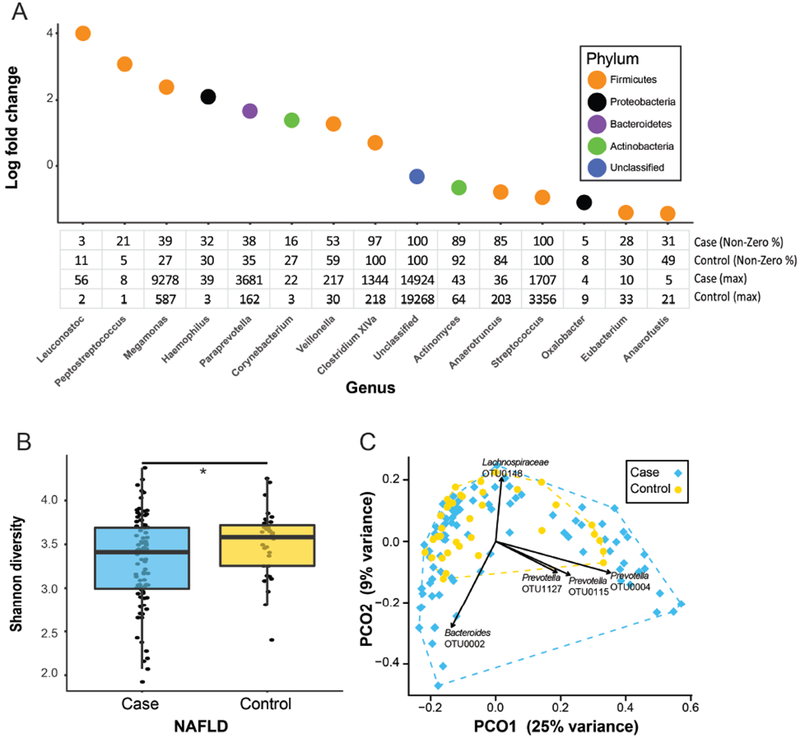
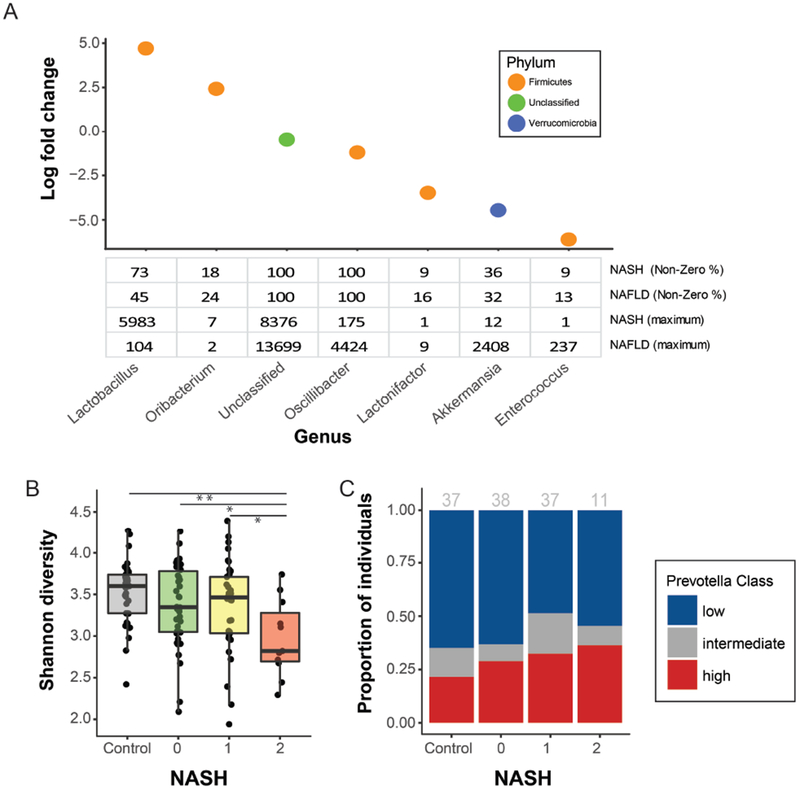
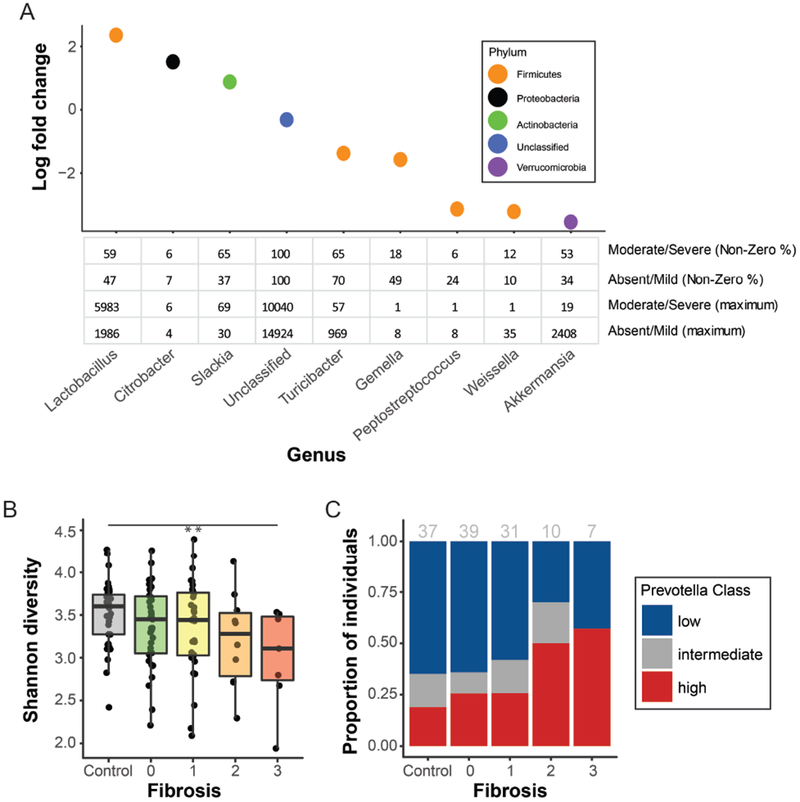
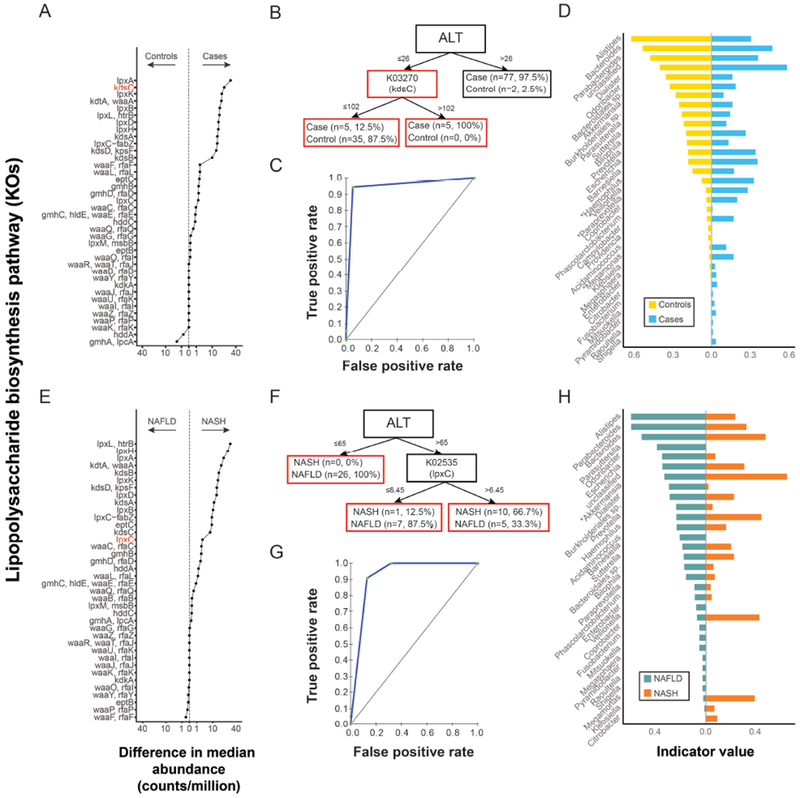
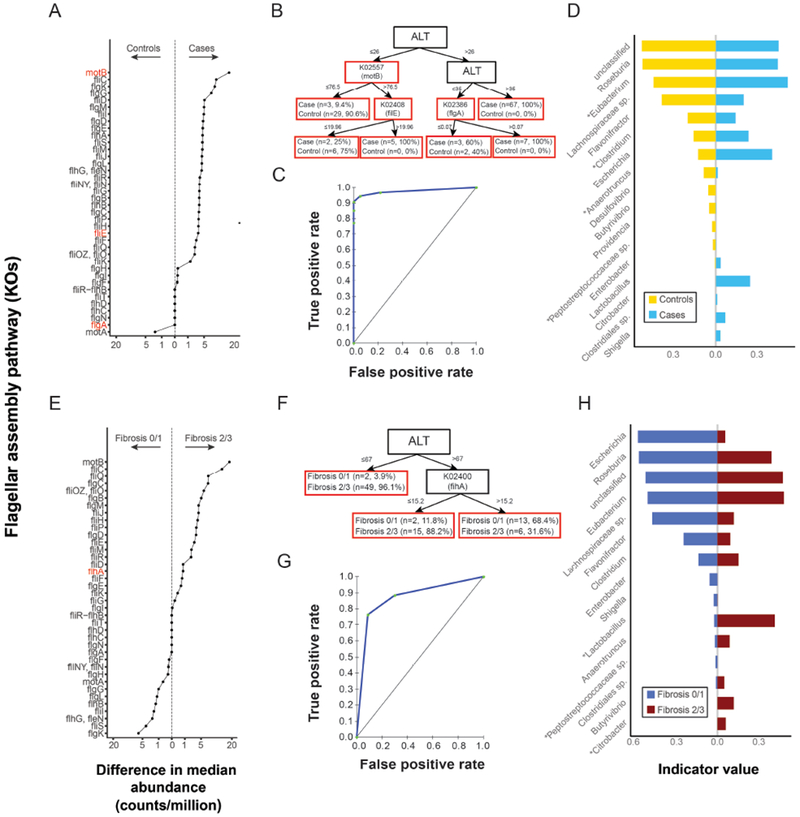
Results: Fecal microbiomes of children with NAFLD had lower α-diversity than those of control children (3.32 vs 3.52, P = .016). Fecal microbiomes from children with nonalcoholic steatohepatitis (NASH) had the lowest α-diversity (control, 3.52; NAFLD, 3.36; borderline NASH, 3.37; NASH, 2.97; P = .001). High abundance of Prevotella copri was associated with more severe fibrosis (P = .036). Genes for lipopolysaccharide biosynthesis were enriched in microbiomes from children with NASH (P < .001). Classification and regression tree model with level of alanine aminotransferase and relative abundance of the lipopolysaccharide pathway gene encoding 3-deoxy-d-manno-octulosonate 8-phosphate-phosphatase identified patients with NASH with an area under the receiver operating characteristic curve value of 0.92. Genes involved in flagellar assembly were enriched in the fecal microbiomes of patients with moderate to severe fibrosis (P < .001). Classification and regression tree models based on level of alanine aminotransferase and abundance of genes encoding flagellar biosynthesis protein had good accuracy for identifying case children with moderate to severe fibrosis (area under the receiver operating characteristic curve, 0.87).
Conclusions: In an analysis of fecal microbiomes of children with NAFLD, we associated NAFLD and NASH with intestinal dysbiosis. NAFLD and its severity were associated with greater abundance of genes encoding inflammatory bacterial products. Alterations to the intestinal microbiome might contribute to the pathogenesis of NAFLD and be used as markers of disease or severity.
Keywords: Flagellin; Intestinal Microbiota; Lipopolysaccharide; Pediatric.
Copyright © 2019 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
There is no conflict of interest to disclose
Figures
Comment in
-
Pediatric NAFLD: lessons from the gut.Hepatobiliary Surg Nutr. 2020 Aug;9(4):534-536. doi: 10.21037/hbsn.2020.01.06. Hepatobiliary Surg Nutr. 2020. PMID: 32832512 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
