

Paralog Studies Augment Gene Discovery: DDX and DHX Genes
- PMID: 31256877
- PMCID: PMC6698803
- DOI: 10.1016/j.ajhg.2019.06.001
Paralog Studies Augment Gene Discovery: DDX and DHX Genes
Abstract
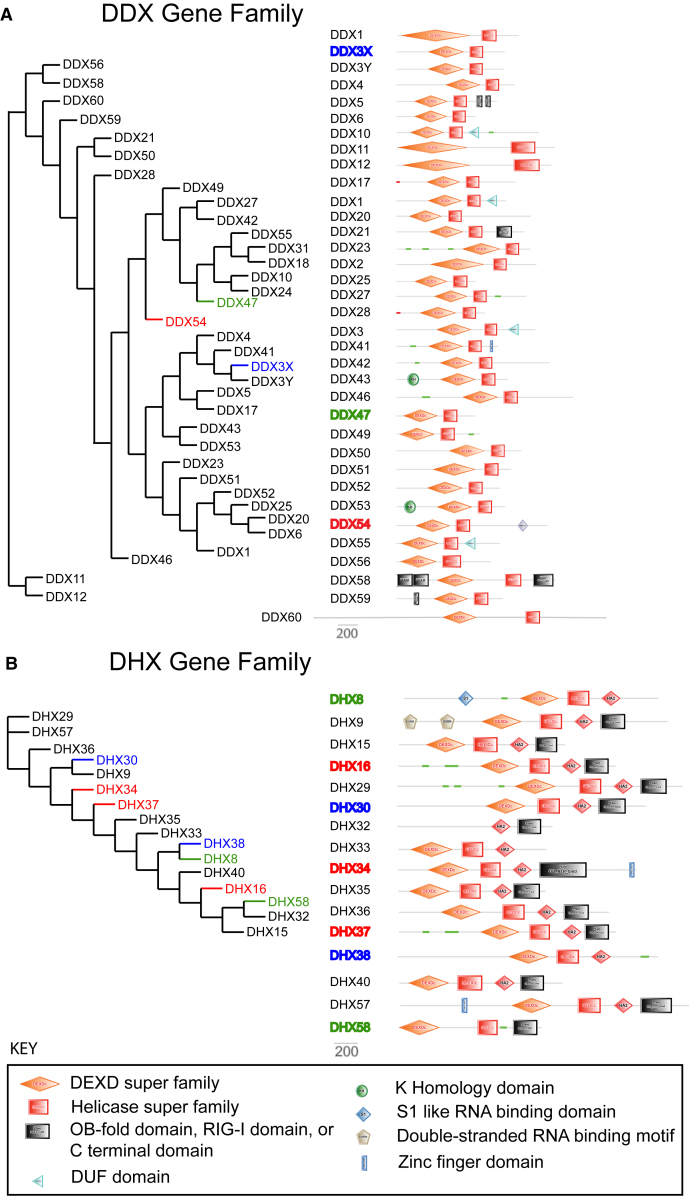
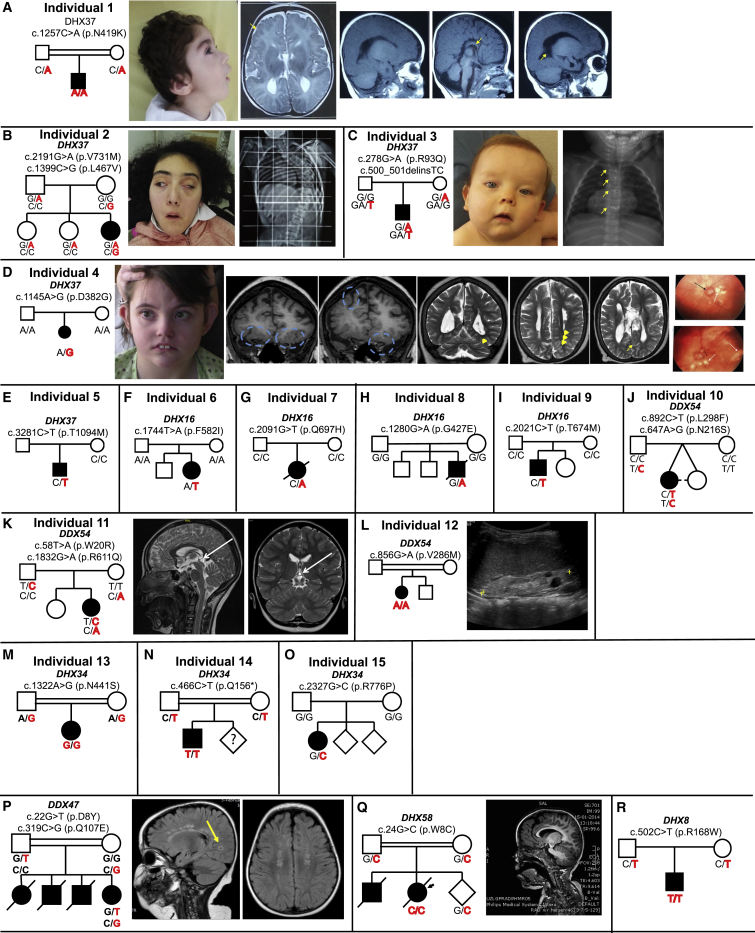
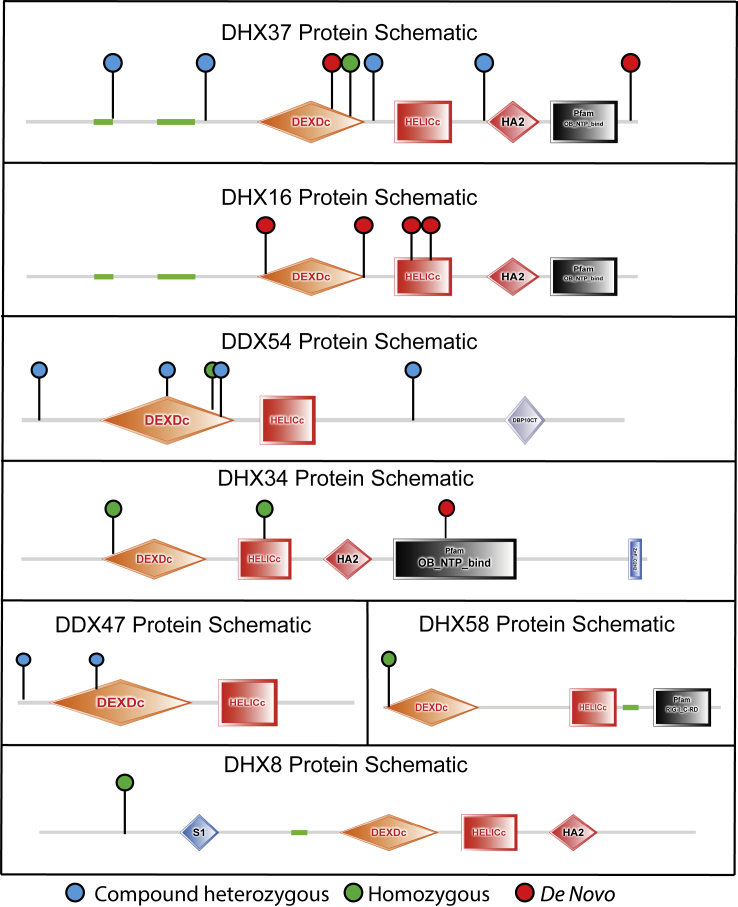
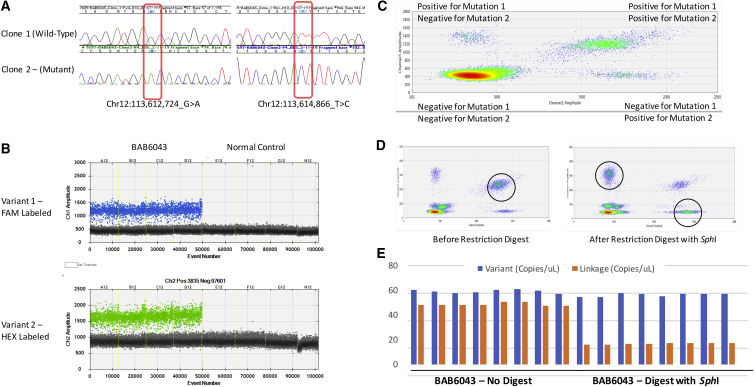
Members of a paralogous gene family in which variation in one gene is known to cause disease are eight times more likely to also be associated with human disease. Recent studies have elucidated DHX30 and DDX3X as genes for which pathogenic variant alleles are involved in neurodevelopmental disorders. We hypothesized that variants in paralogous genes encoding members of the DExD/H-box RNA helicase superfamily might also underlie developmental delay and/or intellectual disability (DD and/or ID) disease phenotypes. Here we describe 15 unrelated individuals who have DD and/or ID, central nervous system (CNS) dysfunction, vertebral anomalies, and dysmorphic features and were found to have probably damaging variants in DExD/H-box RNA helicase genes. In addition, these individuals exhibit a variety of other tissue and organ system involvement including ocular, outer ear, hearing, cardiac, and kidney tissues. Five individuals with homozygous (one), compound-heterozygous (two), or de novo (two) missense variants in DHX37 were identified by exome sequencing. We identified ten total individuals with missense variants in three other DDX/DHX paralogs: DHX16 (four individuals), DDX54 (three individuals), and DHX34 (three individuals). Most identified variants are rare, predicted to be damaging, and occur at conserved amino acid residues. Taken together, these 15 individuals implicate the DExD/H-box helicases in both dominantly and recessively inherited neurodevelopmental phenotypes and highlight the potential for more than one disease mechanism underlying these disorders.
Keywords: DExD/H-box RNA helicase family; developmental delay; human paralogs; intellectual disability.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
J.R.L. serves on the scientific advisory board for Baylor Genetics. J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, and is a coinventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting.
Figures
References
-
- Beighton P., De Paepe A., Steinmann B., Tsipouras P., Wenstrup R.J., Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Am. J. Med. Genet. 1998;77:31–37. - PubMed
-
- Malfait F., Francomano C., Byers P., Belmont J., Berglund B., Black J., Bloom L., Bowen J.M., Brady A.F., Burrows N.P. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C. Semin. Med. Genet. 2017;175:8–26. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
