

BACE1 Translation: At the Crossroads Between Alzheimer's Disease Neurodegeneration and Memory Consolidation
- PMID: 31259308
- PMCID: PMC6597968
- DOI: 10.3233/ADR-180089
BACE1 Translation: At the Crossroads Between Alzheimer's Disease Neurodegeneration and Memory Consolidation
Abstract
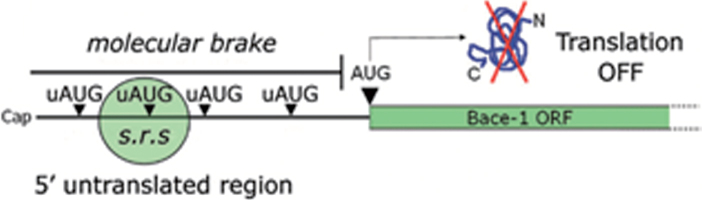
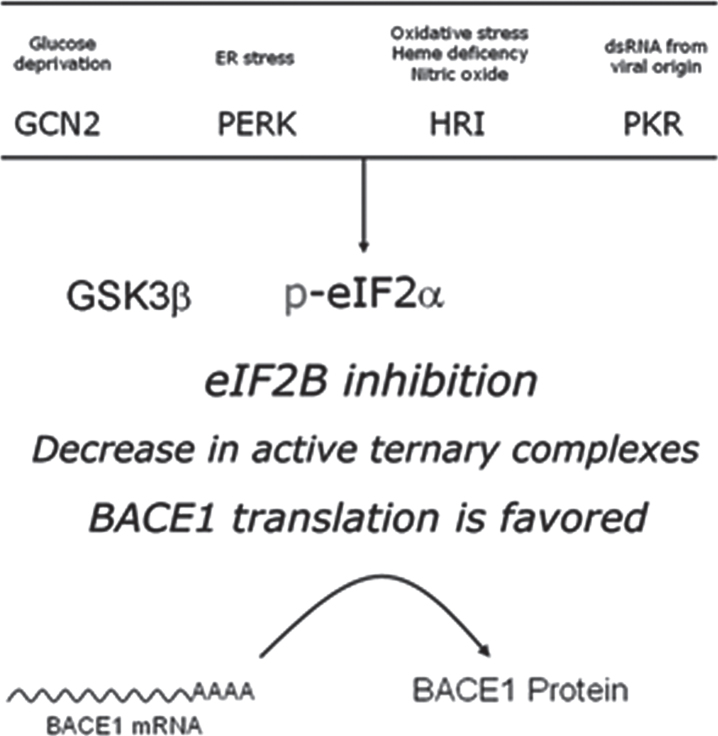
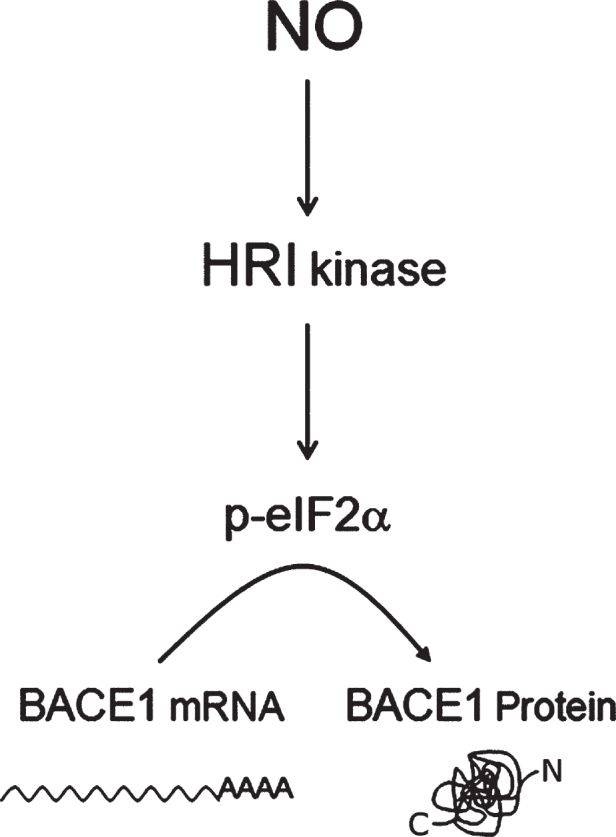
Human life unfolds not only in time and space, but also in the recollection and interweaving of memories. Therefore, individual human identity depends fully on a proper access to the autobiographical memory. Such access is hindered under pathological conditions such as Alzheimer's disease, which affects millions of people worldwide. Unfortunately, no effective cure exists to prevent this disorder, the impact of which will rise alarmingly within the next decades. While Alzheimer's disease is largely considered to be the outcome of amyloid-β (Aβ) peptide accumulation in the brain, conceiving this complex disorder strictly as the result of Aβ-neurotoxicity is perhaps a too straight-line simplification. Instead, complementary to this view, the tableau of molecular disarrangements in the Alzheimer's disease brain may be reflecting, at least in part, a loss of function phenotype in memory processing. Here we take BACE1 translation and degradation as a gateway to study molecular mechanisms putatively involved in the transition between memory and neurodegeneration. BACE1 participates in the excision of Aβ-peptide from its precursor holoprotein, but plays a role in synaptic plasticity too. Its translation is governed by eIF2α phosphorylation: a hub integrating cellular responses to stress, but also a critical switch in memory consolidation. Paralleling these dualities, the eIF2α-kinase HRI has been shown to be a nitric oxide-dependent physiological activator of hippocampal BACE1 translation. Finally, beholding BACE1 as a representative protease active in the CNS, we venture a new perspective on the cellular basis of memory, which may incorporate neurodegeneration in itself as a drift in memory consolidating systems.
Keywords: Alzheimer’s disease; eIF2α; exosomes; heme-regulated eIF-2α kinase; memory; nitric oxide; physiological stress response; proteolysis; translation initiation; β-secretase.
Conflict of interest statement
The authors have no conflict of interest to report.
Figures









Similar articles
-
Physiological Control of Nitric Oxide in Neuronal BACE1 Translation by Heme-Regulated eIF2α Kinase HRI Induces Synaptogenesis.Antioxid Redox Signal. 2015 May 20;22(15):1295-307. doi: 10.1089/ars.2014.6080. Epub 2015 Mar 25. Antioxid Redox Signal. 2015. PMID: 25706765
-
Regulation of Synaptic Amyloid-β Generation through BACE1 Retrograde Transport in a Mouse Model of Alzheimer's Disease.J Neurosci. 2017 Mar 8;37(10):2639-2655. doi: 10.1523/JNEUROSCI.2851-16.2017. Epub 2017 Feb 3. J Neurosci. 2017. PMID: 28159908 Free PMC article.
-
PERK as a hub of multiple pathogenic pathways leading to memory deficits and neurodegeneration in Alzheimer's disease.Brain Res Bull. 2018 Jul;141:72-78. doi: 10.1016/j.brainresbull.2017.08.007. Epub 2017 Aug 10. Brain Res Bull. 2018. PMID: 28804008 Review.
-
Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2α pathway.Biochim Biophys Acta. 2012 Jun;1822(6):885-96. doi: 10.1016/j.bbadis.2012.01.009. Epub 2012 Jan 28. Biochim Biophys Acta. 2012. PMID: 22306812
-
eIF2-dependent translation initiation: Memory consolidation and disruption in Alzheimer's disease.Semin Cell Dev Biol. 2022 May;125:101-109. doi: 10.1016/j.semcdb.2021.07.009. Epub 2021 Jul 23. Semin Cell Dev Biol. 2022. PMID: 34304995 Free PMC article. Review.
Cited by
-
The integrated stress response in brain diseases: A double-edged sword for proteostasis and synapses.Curr Opin Neurobiol. 2024 Aug;87:102886. doi: 10.1016/j.conb.2024.102886. Epub 2024 Jun 19. Curr Opin Neurobiol. 2024. PMID: 38901329 Free PMC article. Review.
-
ARL6IP1 mediates small-molecule-induced alleviation of Alzheimer pathology through FXR1-dependent BACE1 translation initiation.Proc Natl Acad Sci U S A. 2023 May 30;120(22):e2220148120. doi: 10.1073/pnas.2220148120. Epub 2023 May 22. Proc Natl Acad Sci U S A. 2023. PMID: 37216506 Free PMC article.
-
Protein palmitoylation: biological functions, disease, and therapeutic targets.MedComm (2020). 2025 Feb 21;6(3):e70096. doi: 10.1002/mco2.70096. eCollection 2025 Mar. MedComm (2020). 2025. PMID: 39991624 Free PMC article. Review.
-
Berberine Reduces Aβ42 Deposition and Tau Hyperphosphorylation via Ameliorating Endoplasmic Reticulum Stress.Front Pharmacol. 2021 Jul 19;12:640758. doi: 10.3389/fphar.2021.640758. eCollection 2021. Front Pharmacol. 2021. PMID: 34349640 Free PMC article.
-
Unveiling OASIS family as a key player in hypoxia-ischemia cases induced by cocaine using generative adversarial networks.Sci Rep. 2022 Apr 25;12(1):6734. doi: 10.1038/s41598-022-10772-1. Sci Rep. 2022. PMID: 35469040 Free PMC article.
References
-
- Alzheimer A (1907) über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Z Psychiatrie Psychisch-Gerichtliche Med 64, 146–148.
-
- Glenner GG, Wong CW (1984) Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120, 885–890. - PubMed
-
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B (1987) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. - PubMed
-
- Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC (1987) Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science 235, 877–880. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
